¿QUE SON LOS ANTIBIOTICOS?
son sustancia química producida por un ser vivo o derivado
sintético, que mata o impide el crecimiento de ciertas clases de
microorganismo sensibles. Generalmente, son fármacos usados en el
tratamiento de infecciones por bacterias, de allí que se les
conozca como (antibacterianos).
CLASIFICACION DE LOS ANTIBIOTICOS
Estos se clasifican en:
- Biológicos o naturales: Es decir sintetizados por organismos vivos, por ejemplo: la penicilina y el cloranfenicol.
- Semisintéticos: Antibióticos que se obtienen por modificación química de los antibióticos naturales. Por ejemplo: Ampicilina
- Sintéticos: Generados mediante síntesis químicas. Por ejemplo: Sulfas.
VIAS DE ADMINISTRACION
- Por
vía oral: Pueden ser pastillas, cápsulas o líquidos
- Tópicamente:
Puede aplicarse en crema, aerosol o ungüento que se ponga en la piel.
También gotas para los ojos o los oídos
- A través de una inyección o por vía intravenosa: Esto suele utilizarse para infecciones más graves
NO SE DEBEN TOMAR ANTIBIOTICOS PARA:
- Resfriados y
secreción nasal, incluso si la mucosidad es espesa, amarilla o verde
- La
mayoría de los dolores de garganta (excepto la amigdalitis
estreptocócica)
- Gripe
- La mayoría de los casos de bronquitis
¿QUE SON LOS ANTIBIOTICOS SEMISINETICOS?
Algunos antibióticos han
sido sintetizados artificialmente una vez conocida su fórmula estructural,
denominándose sintéticos o semisintéticos. El primer antibiótico obtenido de
esta forma fue el Cloranfenicol en 1949.
Actualmente muchos antibióticos
que originariamente se aislaron a partir de fuentes naturales, hoy en día son
preparados en forma artificial.
De esta manera se ha
mejorado su actividad (espectro), ampliando sus vías de administración y
permite su producción a gran escala.
antibióticos semisintéticos
más importante:
Penicilina:
Ampicilina, alfa
aminobenzil penicilina.
PENICILINA
Las penicilinas son un determinado conjunto de antibióticos con la
capacidad de eliminar las bacterias que causan infecciones en el cuerpo humano.
Estos antibióticos son originados a partir de una particular especie de hongo
conocida como Penicillium y también sirven para prevenir infecciones
bacterianas, especialmente aquellas que son provocadas por las bacterias gran positivas.

CLASIFICACION DE LA PENICILINA
- penicilinas naturales
- penicilinas semisintéticas
- penicilinas sintéticas
PARA QUE SIRVE LA PENICILINA
Penicilina es un anti bactericida. Administrado en dosis suficientes y regulares, sirve para detener una infección de tipo bacteriano o de microorganismos similares, siempre y cuando sean sensibles bioquímicamente a ella penicilina es un anti bactericida.
MECANISMO DE ACCION
Las penicilinas actúan alterando la última fase de la
síntesis de peptidoglucanos de la pared celular, la cual confiere a las
bacterias la resistencia necesaria para soportar la elevada presión osmótica enterobacteriana.
Para alcanzar su punto de acción, las penicilinas tienen que atravesar
distintas estructuras de las bacterias que son diferentes en las Gram positivas
de los gramnegativos. En el primer caso, la pared celular atravesada
fácilmente. En las bacterias gramnegativos el paso del antibiótico se produce
más lentamente y con mayor dificultad, ya que, a diferencia de las anteriores,
en este caso tiene que hacerlo a través de la membrana externa por unos canales
denominados porinas.
¿CUALES SON LAS VENTAJAS Y DESVENTAJAS DE LA PENICILIA?
- Elimina microorganismos patógenos.
- La
bacitracina: Inhibe al transportador lipídico del peptidoglucano hacia el
exterior de la célula.
- La
penicilina: inhibe la transpeptidación, una reacción en la que se producen
los enlaces cruzados de la pared celular.
- Las
cefalosporinas: moléculas que inhiben las proteínas que sintetizan la
pared celular.
PENICILINAS NATURALES
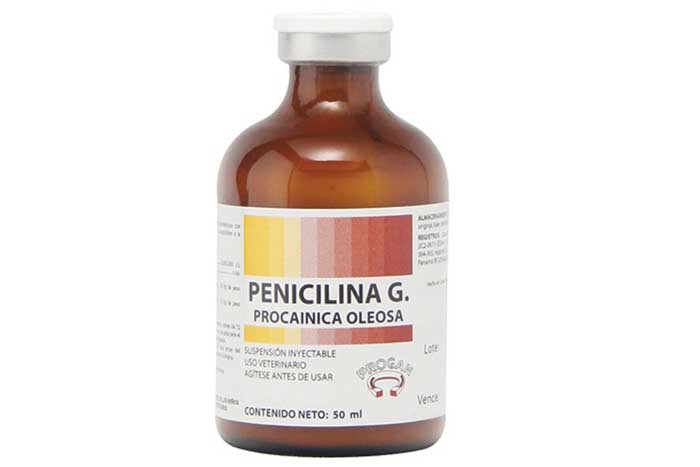
Todas las penicilinas naturales actúan sobre bacterias en multiplicación y al igual que las semisintéticas, tienen que pasar a través de las porinas de la bacteria para unirse a las proteínas fijadoras de penicilina, llamadas PBPs (proteínas fijadoras de penicilina), las cuales están implicadas en la síntesis y maduración de los peptidoglicanos que conforman dicha pared celular.
FARMACOCINETICA
La diferencia entre estas penicilinas es el tiempo de absorción distribución y excreción, no así su vida media que es de 30 a 60 minutos aproximadamente para todas ellas. Así tenemos que la absorción de la penicilina G sódica cristalina es rápida, al igual que su excreción, por lo que hay que administrarla cada 4 a 6 horas por vía intravenosa; la procaína, que tarda más en eliminarse, puede administrarse cada 12 horas y en algunos casos, cada 24 horas, por vía intramuscular; finalmente la penicilina benzatínica, la cual se absorbe lentamente, pero su excreción es muy prolongada, por lo que se administra cada 14 a 21 días, también por vía intramuscular.
FARMACODINAMIA
Absorción vía oral, las penicilinas se deben administrar una hora antes de comer o dos a tres después de comer los alimentos (Niños y Ancianos)
· Distribución: En tejidos y cavidades (liquido pleural, pericardio y sinovial)
· Metabolismo: La mayoría de las penicilinas apenas sufren. Biotransformación en el organismo.
· Eliminación: Tiene lugar primordialmente en él. Riñón por secreción tubular y en casos de insuficiencia renal por las bilis.
La resistencia a la penicilina esta mediada por plásmidos que pueden generar información para producir falla antimicrobiana por varios mecanismos:
El cierre de porinas de la pared bacteriana impide la entrada del antibiótico.
· Por último, la bacteria puede inactivar a la penicilina por medio de betalactamasas excretadas al medio extracelular por grampositivas, o presentes en el espacio periplásmico en las gramnegativo.
PENICILINA G CRISTALINA
Es un antibiótico útil en el tratamiento de enfermedades causadas por
microorganismos sensibles como actinomicosis, ántrax, artritis gonocócica,
bacteriemia por neumococos y estreptococos sensibles; infecciones por
clostridios, difteria activa y prevención del estado de portador; empiema por
neumococos, endocarditis bacteriana, gonorrea, infecciones por Listeria,
meningitis por estreptococos y neumococos sensibles.

REACCIONES ADEVERSAS
·
Náusea.
·
vómitos.
·
Dolor
·
inflamación
·
bultos
·
hemorragia o moretones en el área en donde se
inyectó el medicamento.
INDICACIONES
Está indicado para el tratamiento de infecciones del tracto respiratorio
superior como amigdalitis, faringitis, laringitis; del tracto respiratorio
inferior como la neumonía y bronconeumonía. Infecciones de piel y tejidos
blandos: erisipela, escarlatina, endocarditis, meningitis bacteriana, sífilis,
gonorrea, y en todos los procesos infecciosos causados por bacterias sensibles
a la penicilina.
PENICILINA BENZATINICA
Vía de administración y dosis:
Profilaxis para fiebre reumática y glomerulonefritis: Después del cuadro
agudo se recomienda la administración intramuscular de PENICILINA G BENZATÍNICA
a los niños y adultos en una dosis de 1.2 millones de unidades una vez por mes
o 600,000 unidades cada 2 semanas.
REACCIONES ADVERSAS
·
Náusea
·
Vómitos
·
Dolor
·
Inflamación
·
Bultos
·
hemorragia o moretones en el área en donde se
inyectó el medicamento.
INDICACIONES
La PENICILINA G BENZATÍNICA intramuscular indicada para el tratamiento
de infecciones causadas por microorganismos sensibles a la bencilpenicilina que
sean susceptibles a las concentraciones séricas bajas y muy prolongadas comunes
de esta presentación farmacéutica.
El tratamiento debe ser guiado por los estudios bacteriológicos
(incluyendo pruebas de sensibilidad) y por la respuesta clínica. Las siguientes
infecciones generalmente responderán a la dosis adecuada de PENICILINA G BENZATÍNICA
intramuscular:
·
leves a moderadas de las vías respiratorias
altas.
OXACILINA

FARMACODINAMIA
MECANISMO DE ACCION
Bactericida, con un modo de acción similar al de la bencilpenicilina;
pero es resistente a la penicilinas estafilocócica.
Es activo contra estafilococos productores y no productores de
penicilinas. Su actividad contra estreptococos tales como: Estreptococos
neumonía y Estreptococos piógenas es menor que las bencilpenicilinas, pero
suficiente para ser usado cuando estos microorganismos están presentes con
estafilococos resistentes a la penicilina. Es inefectivo contra Enterococos
faucales.
FARMACOCINETICA
·
Absorción oral: No es completamente absorbida
del tracto gastrointestinal
·
Distribución: Aproximadamente un 93% se une a
proteínas plasmáticas. Atraviesa la barrera
·
Excreción: El fármaco en forma inalterada y sus
metabolitos son excretados en la orina por filtración.
INDICACIONES
Infecciones debidas a estafilococos productores de betalactamasa
incluyendo la otitis externa, coadyuvante en el tratamiento de neumonías,
impétigos, celulitis, osteomielitis y endocarditis estafilocócicas.
·
Frecuentes: diarrea leve, náuseas o vómitos,
fiebre, rash, cansancio o debilidad no habituales y eosinofilia.
·
Ocasionales: neutropenia. Súper infecciones por
organismos resistentes incluyendo la Pseudomona y la Cándida en tratamientos
prolongados, hematuria, eliminación de grandes cantidades de orina de color muy
claro, edema de la cara y lóbulos, respiración dificultosa. Raras: Hepatitis e
ictericia por colestasis.
REACCIONES ADVERSAS
·
Prurigo
·
rash
·
cutáneo
·
urticaria
·
nephritis intersticial
·
diarrea
·
nauseas
·
vómitos.
·
Vía IV a dosis elevadas: convulsiones, toxicidad
en SNC (especialmente en pacientes con fallo renal), flebitis, candidiasis
oral.
CLOXACILINA

MECANISMO DE ACCION
·
Bactericida.
·
Inhibe las transpeptidasas y carboxipeptidasas
impidiendo la síntesis de la pared celular bacteriana.
·
Es resistente a penicilinas estafilocócica.
FARMACOCINETICA
·
Absorción: Tras la inyección intramuscular se
alcanzan concentraciones plasmáticas máximas al cabo de 45 minutos. En adultos,
son aproximadamente de 15 mg/L tras una dosis de 500 mg
DICLOXACILINA

INDICACIONES
La principal indicación es el tratamiento de infecciones por
estafilococo productor de penicilinas, neumococo grupo A-beta, estreptococo
hemolítico y penicilina G-resistentes y penicilina G-estafilococo sensible.
Estudios recientes han reportado que el porcentaje de cepas resistentes de
estafilococo a penicilina G han aumentado en nosocomios, por ello, se
recomienda contra bacterias productoras de penicilinas en terapia inicial.
REACCIONES ADVERSAS
· Produce disturbios gastrointestinales, tal como náusea, vómito, dolor epigástrico, flatulencia, halitosis, ha sido notada en algunos pacientes con el tratamiento de
DICLOXACILINA.
·
Como otras penicilinas produce urticaria,
prurito, rasch cutáneo, eosinofilia, reacción anafiláctica y otros síntomas
alérgicos.
·
Cambios menores en el funcionamiento hepático,
como la elevación de la TGO.
AMOXICILINA
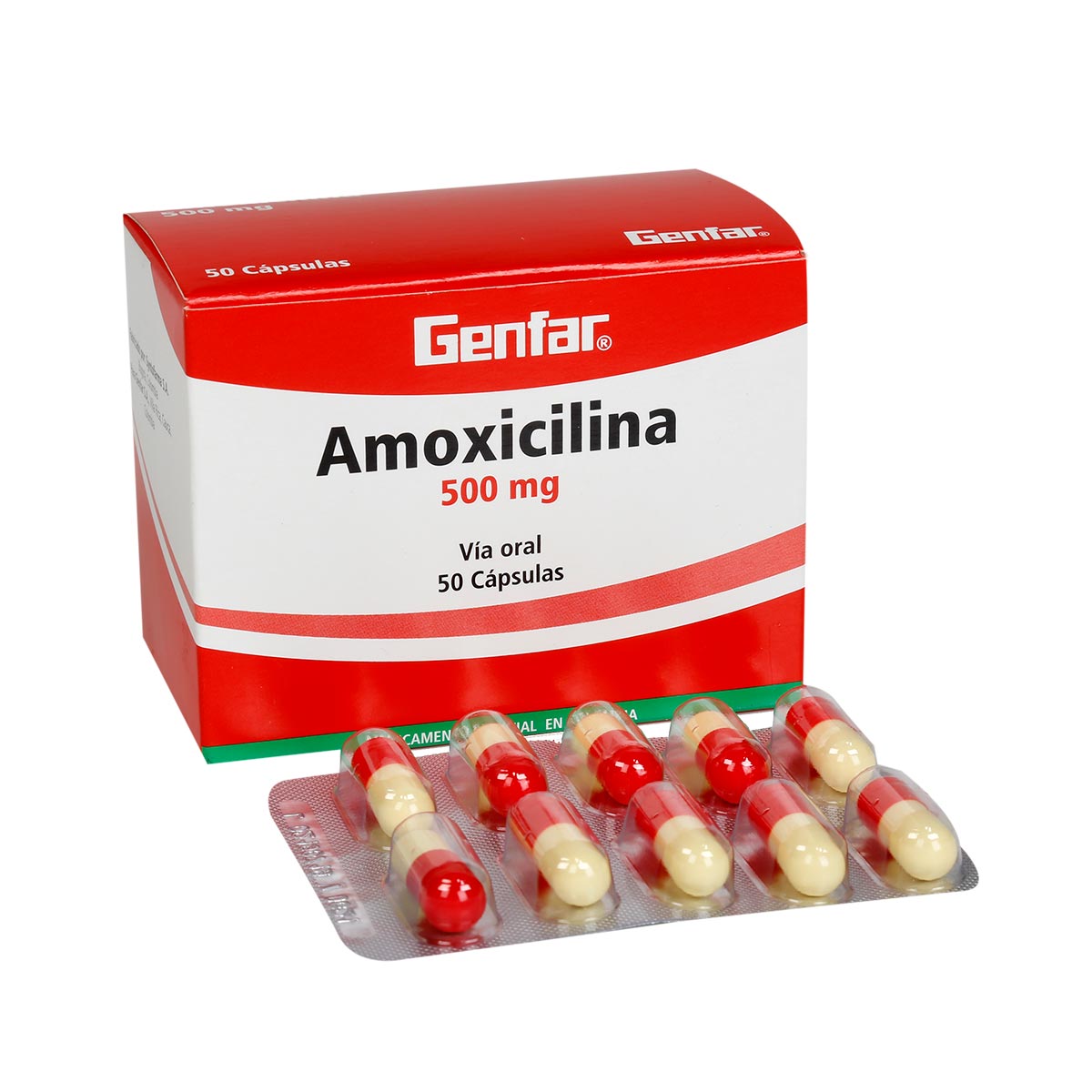
INDICACIONES
La amoxicilina está indicada en el tratamiento de infecciones sistémicas
o localizadas causadas por microorganismos Gram-positivos y Gram-negativos
sensibles, en el aparato respiratorio, tracto gastrointestinal o
genitourinario, de piel y tejidos blandos, neurológicas y
odontoestomatológicas. También está indicado en la enfermedad o beriliosis de
Lyme, en el tratamiento de la infección precoz localizada (primer estadio o
eritema migratorio localizado) y en la infección diseminada o segundo estadio.
REACCIONES ADVERSAS
·
Los efectos secundarios más frecuentes son los
asociados a reacciones de hipersensibilidad y pueden ir desde rash sin
importancia a serias reacciones anafilácticas. Se ha descrito eritema
multiforme, dermatitis exfoliaría, rash maculopapular con eritema, necrólisis
epidérmica tóxica, síndrome de Stevens-Johnson, vasculitis y urticaria.
·
En alguna rara ocasión se observada nefritis
intersticial con necrosis tubular renal y síndrome nefrótico
·
Los efectos secundarios más comunes, asociados
al tracto digestivo son similares a los de otros antibióticos y se deben a la
reducción de la flora: Nausea/vómitos, anorexia, diarrea, gastritis, y dolor
abdominal. En algún caso puede producirse colitis pseudomembranosa durante el
tratamiento o después, si bien este efecto suele ser bastante raro, Pueden
producirse superinfecciones durante un tratamiento con amoxicilina, en
particular si es de larga duración. Se han comunicado candidiasis orales y
vaginales.
AMPICILINA

INDICACIONES
La ampicilina está indicada en el tratamiento de infecciones producidas
por microorganismos sensibles. Unos de los siguientes microorganismos son
considerados sensibles a la ampicilina in vitro: Actinomices sp.; Bacillos atracos;
Bactericidas funduliformis; Bifidobacteria.
REACCIONES ADVERSAS
Se ha demostrado que existe alergenicidad cruzada con otros antibióticos
betalactámicos y carbapenémicos.
En casos raros se ha asociado a la ampicilina con daño tubular renal y
nefritis intersticial, así como eosinofilia, anemia hemolítica y alteración de
las enzimas hepáticas TGO y TGP. Si la función renal se halla restringida, se
debe reducir la dosis de acuerdo con la depuración de creatinina.
Existe la posibilidad de que se presente colitis pseudomembranosa, en
tal caso deberá suspenderse de inmediato la ampicilina.
Localmente se puede presentar en ocasiones tromboflebitis después o
durante la infusión intravenosa, así como dolor local en el sitio de la
inyección intramuscular.
MEZLOCILINA
Es un antibiótico betalactámico bactericida semisintético de amplio
espectro, perteneciente al grupo de las acilureidopenicilinas cíclicas
(alcalina). Su espectro de acción se extiende a un gran número de gérmenes
gramnegativos, grampositivos y anaerobios (Clostridium, Bactericidas).

FARMACOCINETICA
·
Vida media: 60 minutos.
·
Unión a proteínas plasmáticas: 30%.
·
Excreción en orina: luego de la administración
IV, es del 55% al 70% de la dosis dentro de las 24 horas en forma inalterada.
·
Excreción
biliar: luego de administración IV, hasta un 25% de la dosis se excreta en
forma inalterada (activa).
FARMACODINAMIA
La mezlocilina es un antibiótico betalactámico bactericida semisintético
de amplio espectro, perteneciente al grupo de las acilureidopenicilinas
cíclicas similar a la alcalina. Su espectro de acción se extiende a un gran
número de gérmenes gramnegativos, grampositivos y anaerobios, incluyendo cepas
de Clostridium, Bactericidas)
INDICACIONES
Los gérmenes productores de betalactamasas son resistentes a la
mezlocilina. Profilaxis perioperatoria de corto plazo o terapia temprana. Se
recomienda su combinación con otros bactericidas en infecciones sistémicas
graves o por germen desconocido o en infecciones mixtas (por aerobios y
anaerobios).
REACCIONES ADVERSAS
·
Exantemas
·
Prurito
·
Urticaria
·
Otras reacciones alérgicas excepcionales son eosinofilia,
fiebre medicamentosa, nefritis intersticial aguda, vasculitis. Shock
anafiláctico.
PIPERACILINA

MECANISMO DE ACCION
La piperacilina es un
antibiótico beta-lactantico, principalmente bactericida. Inhibe la tercera y
última etapa de la síntesis de la pared celular bacteriana uniéndose
preferentemente a las proteínas de unión a penicilinas (PBP específicas) que se
encuentran dentro de la pared celular bacteriana.
FARMACOCINETICA
Se administra por vía intravenosa en infusión 30 minutos. La media de
las concentraciones plasmáticas máximas de piperacilina son aproximadamente
134, 242, y 298 mg/ml después de dosis de 2,25 g, 3,375 g, y 4,5 g.
INDICACIONES
Tratamiento de infecciones producidas por bacilos aerobios
Como con todas las penicilinas, puede ocurrir toxicidad sobre el SNC con
dosis IV masivas o con exceso de dosis en pacientes con función renal dañada;
caracterizada por confusión, somnolencia, y mioclonía que puede progresar hasta
convulsiones y acabar en muerte.
Pueden producirse
alteraciones inmunológicas como enfermedad del suero y reacciones
anafilácticas.
Alteraciones sanguíneas como eosinofilia (2-8%) y, rara vez, aumento del
tiempo de protrombina, hemorragias, anemia hemolítica, neutropenia, leucopenia,
trombocitopenia, y púrpura.
Rara vez aparece hematuria o nefritis intersticial
CEFALOSPORINAS
Las cefalosporinas son antibióticos similares a las penicilinas, pero
resultan más efectivas porque han mostrado tener una mejor resistencia contra
las B-lacta masas.
CEFALOSPORINAS DE 1RA GENERACION
Es la más antigua de las cefalosporinas parenterales de primera
generación. Es activa contra la mayoría de los cocos Gram positivos, incluyendo
a los estafilococos productores de penicilinas, pero no tiene actividad contra
entero cocos, Listeria, estafilococos resistentes a oxacilina y neumococos
resistentes a penicilina. Es activa contra la mayoría de cepas de E. coli,
Proteus mira bilis y neumonía, pero tiene una débil actividad contra Proteus
indol positivo.
CEFADROXILO

MECANISMO DE ACCION
Bactericida, antibiótico
de amplio espectro Inhibe la síntesis y reparación de la bacteriana
INDICACIONES
·
Infecciones del
tracto urinario, causadas por Escherichia coli, P. Mira bilis y especies у de
Klebsiella.
·
Infecciones de
la piel y estructuras relacionadas causadas por estreptococos y estafilococos.
·
Faringitis amigdalitis
causadas por estreptococo beta hemolítico Grupo A
FARMACOCINETICA
El medicamento es
rápidamente excretado por el riñón; más de un 85 % aparece inalterado en la
orina dentro de las 8 primeras horas, la mayor parte dentro de las 2 primeras
horas. Durante este periodo de 8 horas, las concentraciones urinarias máximas
tras dosis de 250 mg, 500 mg y 19. Fueron aproximadamente de 600, 900 y 1900
mg/l respectivamente.
FARMACODINAMIA
antibacteriano
betalactámico semisintético de amplio espectro, cefalosporinas de primera generación.
Estos fármacos El cefadroxilo es un perteneciente a la familia de las actúan
inhibiendo la síntesis de la pared bacteriana durante la multiplicación
interfiriendo con la fase final de la síntesis de peptidoglicano. Posee
actividad bactericida.
INTERVENCIONES DE ENFERMERIA
·
Orientar al paciente
acerca de medicamento a administrar.
·
Observar si se
presentan reacciones de hipersensibilidad y reportar al médico.
·
Respetar el
intervalo de administración recomendado.
EFECTOS SECUNDARIOS
·
Malestar estomacal
·
Diarrea
·
Vómitos
·
Sarpullido leve
·
Purito
·
Urinarias
·
Resoplo (silbido al respirar)
REACCIONES ADVERSAS
·
Colitis pseudomembranosa
·
Neutropenia moderada
·
Anafilaxis Ictericia colestásica
PRECAUCIONES
Administrar con precaución en pacientes con hipersensibilidad a penicilinas
y modificar dosis en pacientes con disminución de la función renal. No usar en
pacientes con reacción de hipersensibilidad inmediata a la penicilina. La administración
prolongada de cefadroxilo podría resultar en sobreinfección bacteriana o fúngica
(candidiasis).
CEFALOSPORINAS DE SEDUNDA GENERACION
Ofrecen una cobertura
mayor frente a los bacilos gramnegativos que las de primera generación. Todas
estas cefalosporinas tienen actividad contra la mayoría de los microorganismos
destruidos por agentes de la primera generación, pero su cobertura es más
extensa ya que incrementan su actividad contra microorganismos gran-negativos,
en especial dos: Haemophilus influenzae y Neisseria sp.
CEFUROXIMA

MECANISMO DE ACCION
Cefuroxima (como axetilo) sufre hidrólisis mediante las enzimas esterasas
para transformarse en el antibiótico activo, cefuroxima. Cefuroxima inhibe la
síntesis de la pared celular bacteriana después de la unión a las proteínas
fijadoras de penicilina Penicilina... Binding Proteins, PBPs). Esto tiene como
resultado la interrupción de la biosíntesis de la pared celular (peptidoglicano)
lo que produce la lisis celular bacteriana y muerte.
INDICACIONES
·
Amigdalitis estreptocócica aguda y faringitis.
·
Sinusitis bacteriana aguda.
·
Otitis media aguda
·
Exacerbación aguda de la bronquitis crónica.
·
Cistitis
·
Pielonefritis.
·
Infecciones no complicadas de: la piel y tejidos
blandos.
·
Tratamiento de las primeras fases de la
enfermedad de Lyme
FARMACOCINETICA
la cefuroxima se administra parenteralmente en forma de sal sódica y
oralmente en forma de axetil. Después de una dosis intramuscular los niveles
máximos en plasma se alcanzan en 15-60 minutos. Por su parte, la cefuroxima
axetil: es rápidamente hidrolizada en la mucosa intestinal. penetrando entre
el. 37 y 52% de la dosis en la circulación sistémica Después de una administración
oral, los niveles máximos de cefuroxima se alcanzan en las dos primeras horas. Aproximadamente
el 35 50% de la cefuroxima circulante se...encuentra unida a las proteínas del plasma
FARMACODINAMIA
La cefuroxima es muy activa frente a la mayoría de las bacterias gran-positivas
(incluyendo las cepas productoras de penicilinasa) como los estafilococos (S. áureas,
S. epidermis), estreptococos (a excepción de los enterococos) y algunas
bacterias: gran positivas anaerobias. Entre las bacterias negativas sensibles a
la cefuroxima se encuentran los E. coli. Klebsiella, H. influenza. Proteus mira
bilis. N. meningitis, y N. gonorrhoeae, incluyendo las cepas que son
productoras de beta lacta masas.
INTERVENCIONES DE ENFERMERIA
·
Antes de comenzar el tratamiento, se debe
establecer si el paciente tiene antecedentes de reacciones de hipersensibilidad
graves a cefuroxima.
·
Vigilar si presentas se reacciones adversas.
·
No incurrir situaciones en innecesarias por
falta de atención o conocimiento
EFECTOS SECUNDARIOS
·
Diarrea
·
Nauseas
·
Dolor abdominal
·
Cefaleas
·
Mareos
REACCIONES ADVERSAS
·
Eosinofilia
·
Colitis
·
pseudomembranosa
·
Anafilaxia
·
Aumento transitorio de los niveles de las
enzimas hepáticas
·
Tromboflebitis
PRECAUCIONES
Hipersensibilidad a otros betalactámicos.
Se deben administrar con precaución a pacientes que reciben tratamiento
simultáneo con diuréticos potentes
RECOMENDACIONES
No aplicar más de 1 g en un mismo punto, cuando se usa vía IM
CEFALOSPORINAS DE TERCERA GENERACION
Las cefalosporinas de
tercera generación son el tratamiento de elección en la meningitis por bacilos
gramnegativos y se utilizan también para combatir otras infecciones por bacilos
gramnegativos. Al igual que en los grupos anteriores hay parenterales y orales.
CEFALOSPORINAS DE CUARTA GENERACION
Tienen un mayor espectro
contra organismos gram-positivos que las cefalosporinas de la tercera
generación. También tienen una mayor resistencia a beta-lactamasas que las
cefalosporinas de la tercera generación. Cubren Enterobacter y Citrobacter y
tiene particular uso terapéutico en el tratamiento de infecciones debidas a
bacilos aerobios gram-negativos resistentes a cefalosporinas de tercera
generación con lo cual se logra su erradicación y tienen mejor actividad contra
algunos gram-positivos.
CEFALOSPORINAS DE QUINTA GENERACION
Ceftobiprol es una
cefalosporina activa tanto frente a Staphylococcus aureus como
estafilococos coagulasa negativos, sensible y resistente a meticillina, así
como frente a las cepas resistentes a vancomicina (VRSA).
FAMACOCINETICA
Todas las cefalosporinas a
excepción de la cefalexina, cefadroxilo, cefradina, cefaclor, cefuroxime
axetil, cefprozil, cefixime, cefpodoxima proxetil, ceftibuten y cefdinir, deben
ser administradas parenteralmente porque tienen una pobre absorción oral y no
alcanzan concentraciones sistémicas adecuadas para el tratamiento.
USOS
En cuanto a sus usos
terapéuticos estos son muy variados, son efectivas tanto para el tratamiento y
profilaxis de infecciones producidas por bacterias sensibles a su efecto
antibacteriano. Pueden ser usadas como alternativa para las penicilinas en una
variedad de infecciones en pacientes que no pueden recibir penicilinas. Estas
infecciones incluyen aquellas causadas
por Staphylococcus y Streptococcus.
EFECTOS ADVERSOS
Los efectos secundarios
que se pueden observar con las cefalosporinas son principalmente las reacciones
de hipersensibilidad, entre las reacciones inmediatas se encuentran la
anafilaxis, la urticaria y el broncoespasmo.
Las reacciones tardías
incluyen dermatitis, lesiones de la mucosa oral, fiebre y erupciones cutáneas.
Lo más común es que se
presente una erupción maculopapular, habitualmente durante el tratamiento, la
cual puede en ciertas ocasiones acompañarse de fiebre y eosinofilia. Pueden
producir reacciones locales en la zona de la inyección intramuscular, además de
flebitis y tromboflebitis cuando se utiliza la vía intravenosa. Puede aparecer
náuseas, vómitos, dolor abdominal, tienden a producir en raras ocasiones
depresión de la médula ósea, caracterizada por granulocitopenia.
PRECAUCIONES EN EL USO
·
Alergias
·
Diabetes
·
Embarazo
·
Lactancia
materna
FARMACOS ANTIARRÍTMICOS
Los
fármacos antiarrítmicos forman un grupo muy heterogéneo de sustancias que se
caracterizan por suprimir o prevenir las alteraciones del ritmo cardíaco a
concentraciones a las que no ejercen efectos adversos sobre el latido sinusal normalmente
propagado. En la actualidad, continúan siendo el tratamiento de elección en la
mayoría de los pacientes con arritmias, aunque diversas estrategias eléctricas
(desfibriladores, marcapasos y técnicas de ablación) y quirúrgicas pueden
reemplazarlos en determinados grupos de pacientes. Las alteraciones del ritmo
cardíaco son el resultado de anomalías en: a) la génesis del impulso cardíaco
(alteraciones del automatismo), y b) la secuencia de activación del miocardio
(alteraciones de la conducción o reentrada). Estas anomalías del automatismo o
de la conducción del impulso cardíaco pueden ser desencadenadas bien por
cambios en los mecanismos iónicos responsables de la génesis o el mantenimiento
de los potenciales de acción cardíacos; bien por alteraciones de tipo
anatómico-funcional (p. ej., cardiopatía isquémica, hipertrofia ventricular o
fibrosis). En este capítulo se explicarán los conceptos básicos de la
electrofisiología cardíaca del miocardio sano y enfermo, los mecanismos
desencadenantes de las arritmias cardíacas y, por último, la farmacología de
los agentes antiarrítmicos utilizados actualmente en la prevención y supresión
de las arritmias cardíacas.
CLASIFICACION DE LOS FARMACOS ANTIARRITMICOS
Clásicamente,
los fármacos antiarrítmicos han sido divididos en cuatro grupos (tabla 38-1):
Grupo I: fármacos cuyo mecanismo de acción es el bloqueo de los canales de Na+
dependientes del voltaje. Los fármacos pertenecientes a este grupo inhiben la
INa y, por lo tanto, disminuyen la velocidad de conducción y la excitabilidad
cardíacas. Grupo II: fármacos que actúan bloqueando receptores b-adrenérgicos.
Grupo III: fármacos cuyo mecanismo de acción es producir una prolongación de la
duración del potencial de acción y, por lo tanto, del período refractario.
Grupo IV: fármacos bloqueantes de los canales de Ca2+ dependientes del voltaje
de tipo L, con la excepción de las dihidropiridinas. Al inhibir la ICa,
disminuirán la velocidad de conducción y el período refractario de las células
cardíacas de los nodos SA y AV, así como de células cardíacas anormalmente
despolarizadas (p. ej., células de miocardio isquémico). Aunque esta
clasificación es sencilla, clara y fácil de recordar, presenta numerosos
inconvenientes. El primero es que mezcla efectos bloqueantes de canales y
receptores con la modificación de un parámetro electrofisiológico (duración del
potencial de acción). Además, no contempla la posibilidad de que el aumento de
una determinada corriente iónica pueda ejercer efectos antiarrítmicos, ni que
haya bloqueo ni estimulación de otro tipo de receptores distintos de los
b-adrenérgicos, por lo que diversos fármacos con actividad antiarrítmica
(adenosina, bloqueantes a-adrenérgicos, digoxina, atropina, etc.) quedan
excluidos de la clasificación. Asume que el mecanismo de acción de los fármacos
pertenecientes a un mismo grupo está únicamente relacionado con el descrito en
la clasificación; sin embargo, la mayor parte de los fármacos antiarrítmicos
comparten más de un mecanismo de acción antiarrítmico. Esto explica por qué, en
contra de lo que al parecer indica esta clasificación, antiarrítmicos incluidos
en un mismo grupo presentan propiedades y aplicaciones clínicas muy dispares.
Las tablas 38-2 y 38-3 muestran las características electrofisiológicas y el
espectro antiarrítmico de los principales antiarrítmicos.
FARMACOS
ANTIARRITCICOS DEL GRUPO 1
MECANISMOS
GENERALES DE ACCION
Como
ya se ha mencionado, el mecanismo de acción común a los fármacos antiarrítmicos
del grupo I es el bloqueo de los canales de Na+ dependientes del voltaje. Este
efecto produce, como consecuencia, una disminución de la excitabilidad y de la
velocidad de conducción intracardiaca. Esta disminución de la velocidad de
conducción es capaz de suprimir las arritmias por reentrada, ya que el área de
bloqueo unidireccional se convertirá en un área de bloqueo bidireccional. El
mecanismo de acción de los fármacos antiarrítmicos del grupo I se explica
mediante la hipótesis del receptor modulado, propuesta en 1977 por Hondeghem y
Katzung, y que se ilustra en la figura 38-8. Esta hipótesis propone que: a) los
canales de Na+ pueden estar en tres estados diferentes: reposo (estado
cerrado), que predomina a potenciales de membrana electronegativos, abierto
(único estado conductor del canal), que aparece durante la fase 0 del potencial
de acción, e inactivo (estado cerrado no disponible para ser activado), que
predomina a potenciales de membrana electropositivos (durante las fases 2 y 3
del potencial de acción, así como al comienzo de la fase 4); b) los fármacos
antiarrítmicos pueden unirse a cualquiera de los tres estados del canal, aunque
a concentraciones terapéuticas presentan, por lo general, muy baja afinidad por
el estado de reposo y muy alta afinidad por los estados abierto y/o inactivo
del canal; c) los canales de Na+ unidos a fármaco no son conductores (no dejan
pasar iones Na+ a su través), sea cual sea el estado en que se encuentren, y d)
las transiciones entre los canales de Na+ están regidas por una función que
depende del nivel del potencial de membrana y del tiempo. Con un antiarrítmico,
esta función está desplazada hacia niveles más negativos de potencial de
membrana (el paso desde el estado inactivo hasta el de reposo requiere mayor
hiperpolarización que en ausencia de fármaco) y está retrasada en el tiempo (el
tren pasa de 25 msega 0,3-15 seg).
Según
los valores de la tre, los antiarrítmicos del grupo I han sido subdivididos en
tres subgrupos Subgrupo IC: fármacos con una cinética lenta de recuperación del
bloqueo (tre mayor de 8 seg). Dado que el tiempo que tardan en disociarse del
canal es mucho mayor que el intervalo diastólico en ritmo sinusal (1 seg),
estos fármacos producen una marcada depresión de la INa y de la velocidad de
conducción intracardiaca, prolongando el QRS incluso en pacientes en ritmo
sinusal. Está marcada depresión de la velocidad de conducción facilita la
aparición de arritmias por reentrada y sería responsable de la alta incidencia
de fenómenos arritmogénicos que estos fármacos producen. En general, los
fármacos antiarrítmicos pertenecientes a los subgrupos IA y IC presentan muy
alta afinidad por el estado abierto del canal de Na+ , mientras que los
pertenecientes al subgrupo IB exhiben mayor afinidad por el estado inactivo de
aquél. En todos los casos, la afinidad que presentan por el estado de reposo es
muy baja, tal y como ya se ha mencionado. Los fármacos antiarrítmicos del grupo
I que se unen preferentemente al estado abierto del canal de Na+ deprimirán
tanto más la INa cuanto mayor sea la frecuencia de la taquicardia (bloqueo
frecuencia dependiente), ya que, en estas condiciones, el canal pasa más veces
en la unidad de tiempo por el estado abierto (fig. 38-8). Además, y puesto que
el tiempo que el canal se encuentra en estado abierto es el mismo para todos
los tejidos cardíacos (aurícula o ventrículo), estos fármacos serán igualmente
efectivos en el tratamiento de taquiarritmias supraventriculares y
ventriculares.
FÁRMACOS ANTIARRÍTMICOS DEL GRUPO IA
QUINIDINA
Es
el d-enantiómero del antipalúdico quinina, alcaloide obtenido de la corteza de
Cincona, que bloquea el estado activo del canal de Na+ y diversas corrientes de
salida de K+ , a la vez que presenta importantes acciones antimuscarínicas.

ACCIONES
FARMACOLOGICAS
Por
su capacidad para inhibir la INa, la quinidina disminuye la excitabilidad y la
velocidad de conducción intraauricular e intraventricular, por lo que ensancha
el complejo QRS del ECG; este ensanchamiento es un signo temprano de toxicidad
y sirve para monitorizar su dosificación. A la altura del circuito de
reentrada, la depresión de la velocidad de conducción intracardíaca podría
convertir el área de bloqueo unidireccional en bidireccional, suprimiendo la
recirculación del impulso cardíaco. La depresión de la conducción intracardíaca
depende de los niveles plasmáticos de K+ , de tal forma que la hipopotasemia
antagoniza los efectos de la quinidina, mientras que la hiperpotasemia los
potencia. La quinidina bloquea también diversas corrientes de salida de K+, lo
que explica por qué prolonga la duración del potencial de acción (prolonga los
intervalos QTc y JT del ECG) y del período refractario auricular y ventricular.
La quinidina deprime por acción directa el
automatismo del nodo SA y suprime la actividad automática del sistema de
His-Purkinje, tanto por deprimir la inclinación de la fase 4 como por desplazar
el potencial umbral hacia valores menos negativos (tabla 38-2). Sin embargo, no
suprime el automatismo anormal o los pospotenciales tardíos. También deprime la
conducción a través del nodo AV (prolonga el intervalo PR del ECG), así como la
contractilidad y el volumen minuto cardíacos. Sin embargo, a dosis terapéuticas
estas acciones cardio depresoras directas de la quinidina sobre los nodos SA y
AV son contrarrestadas por sus acciones antimuscarínicas y por el aumento
reflejo del tono simpático que su acción vasodilatadora produce. Ello explica
por qué a dosis terapéuticas la quinidina no disminuye, o incluso aumenta, la
frecuencia sinusal y en pacientes con flúter o fibrilación auricular la
quinidina puede acelerar la conducción a través del nodo AV y aumentar
bruscamente la frecuencia ventricular. Para evitar este riesgo, se recomienda
digitalizar al paciente antes de administrar la quinidina. A nivel vascular,
produce vasodilatación e hipotensión tanto por una acción directa sobre la
fibra muscular lisa vascular como por sus propiedades bloqueantes
a-adrenérgicas. La hipotensión, que aparece con mayor frecuencia tras su
administración por vía IV, aumenta por vía refleja el tono simpático, lo que
unido a su acción antimuscarínica oculta parcialmente su acción depresora de la
contractilidad cardíaca. Esta depresión no suele ser un problema en pacientes
con función ventricular normal, pero en pacientes con depresión previa puede
conducir a una insuficiencia cardíaca. A altas dosis, la vasodilatación
periférica unida a la depresión de la contractilidad y del volumen minuto
explicarían la aparición del síncope quinidínico.
La
quinidina se absorbe bien por vía oral, alcanzándose concentraciones
plasmáticas máximas al cabo de 1- 2 horas con el sulfato y 3-4 horas con el
gluconato. También se puede administrar por vía IV, pero no por vía IM, ya que
es irritante y produce dolor local. Se une en el 80 % a proteínas plasmáticas y
se distribuye ampliamente, pero no atraviesa la barrera hematoencefálica. El 90
% de la dosis administrada se hidroxila en el hígado en metabolitos activos
(2-hidroxiquinidina y dihidroxiquinidina), que se eliminan por vía renal; el 10
% restante se elimina por vía renal sin biotransformar. La eliminación
disminuye en pacientes con insuficiencia hepática, cardíaca o renal y en
ancianos, por lo que en estas circunstancias se reducirá la dosis de
mantenimiento. Su semivida es muy variable (3-16 horas), lo que exige
individualizar la dosis utilizando criterios clínicos (ECG y efectos sobre la
arritmia) o determinando sus niveles plasmáticos. La eliminación renal de la
quinidina (pKa = 8,0) aumenta al acidificar la orina y por diálisis.
REACCIONES
ADVERSAS
Las
más comunes son las digestivas (diarrea, anorexia, náuseas o vómitos) y las
anticolinérgicas (sequedad de boca, estreñimiento, visión borrosa o retención
urinaria), que obligan a suspender el tratamiento hasta en el 20 % de los
pacientes. Estas acciones anticolinérgicas contraindican su uso en pacientes
con glaucoma, hipertrofia de próstata o miastenia grave. También produce
reacciones de hipersensibilidad: fiebre, urticaria, asma, trombocitopenia y
hepatitis. A dosis elevadas puede producir: a) cinconismo, un síndrome
caracterizado por cefaleas, acufenos, dificultad de audición, vértigo,
alteraciones visuales (visión borrosa, diplopía o alteración de la percepción
de los colores), confusión, alucinaciones y psicosis, y b) reacciones adversas
cardiovasculares: hipotensión grave con riesgo de colapso, bloqueos AV e
intraventriculares, bradicardia, depresión de la contractilidad y taquicardias
polimórficas ventriculares (torsades de pointes). La aparición de efectos
arritmogénicos ventriculares aumenta cuando la quinidina se administra por vía
IV, así como en pacientes con hipopotasemia o bradicardia y suelen ir
precedidos de un ensanchamiento del QRS por encima del 40 % de su valor y del
intervalo QT del ECG corregido para la frecuencia cardíaca (QTc > 440 mseg).
PROCAINAMIDA
Es un derivado del anestésico local procaína
(figura 38-9) con propiedades similares a las de la quinidina (tablas 38-2 y
38-3). Sin embargo, presenta menor acción antimuscarínica, lo que explica por
qué produce mayor efecto depresor de la frecuencia sinusal y de la conducción
AV que la quinidina. También presenta propiedades bloqueantes ganglionares y
vasodilatadoras directas que son responsables de la hipotensión que aparece
particularmente cuando se utiliza por vía IV.

FARMACOCINETICA
Se
absorbe bien por vía oral, alcanzando su efecto máximo en 1- 2 horas (tabla
38-4). La absorción oral disminuye tras un infarto de miocardio, por lo que en
este caso será administrada por vía IV, alcanzando su efecto máximo en 15-30
min. Se une poco a proteínas plasmáticas (20 %) y se distribuye ampliamente,
aunque no atraviesa la barrera hematoencefálica. El 30-50 % de la dosis se
acetila en el hígado a N-acetil procainamida (NAPA) que prolonga la duración
del potencial de acción (intervalo QT del ECG), es decir, presenta propiedades
antiarrítmicas del grupo III. La velocidad de biotransformación muestra una
distribución bimodal, siendo mayor la formación de NAPA en los acetiles adores
rápidos que en los lentos. Procainamida y NAPA se eliminan rápidamente por vía
renal, acelerándose este proceso al acidificar la orina. La semivida de la
procainamida aumenta de 3,5 a 6-8 horas (de 6-8 hasta 100 horas para la NAPA)
en pacientes con insuficiencia renal o cardíaca (disminuye el volumen de
distribución y el flujo renal), por lo que en estos grupos de pacientes se
recomienda reducir la dosis al menos el 25 %.
REACCIONES ADVERSAS
Las
digestivas y anticolinérgicas son similares, aunque menos frecuentes que las
que aparecen tras la administración de quinidina, pero produce con mayor
frecuencia reacciones de hipersensibilidad (fiebre, urticaria, angioedema y
agranulocitosis), particularmente cuando se utilizan preparaciones de acción
retardada. A dosis altas, casi el 80 % de los pacientes desarrolla al cabo de 6
meses de tratamiento anticuerpos antinucleares y el 20 % desarrolla un síndrome
similar al lupus eritematoso que cursa con fiebre, artralgias, eritemas,
hepatomegalia, pericarditis y dolor pleurítico. Este cuadro persiste semanas o
meses tras suspender el tratamiento y es más frecuente en los acetiles adores
lentos. A dosis altas, produce efectos indeseables: a) neurológicos (mareos,
temblor, debilidad, vértigo, psicosis, alucinaciones, depresión y confusión) y
b) cardiovasculares: hipotensión con riesgo de colapso cuando se administra por
vía IV, bradicardia, bloqueo AV, depresión de la contractilidad y efectos
arritmogénicos (torsades de pointes). El riesgo de aparición de efectos
arritmogénicos es más elevado cuando existe una prolongación de los intervalos
QRS y QT del ECG, y en pacientes con insuficiencia renal en los que se acumula
la NAPA. La procainamida está contraindicada en pacientes con bloqueo AV,
alteraciones de la conducción intraventricular, bradicardia, insuficiencia renal
o cardíaca e hipersensibilidad al fármaco.
Cimetidina
y ranitidina inhiben su biotransformación y aumentan sus niveles plasmáticos,
mientras que el alcohol acelera su eliminación. La procainamida potencia el
bloqueo neuromuscular producido por los antibióticos aminoglucósidos.
DISOPIRAMIDA
Sus propiedades electrofisiológicas son
similares a las de la quinidina, aunque presenta propiedades antimuscarínicas e
inotrópicas negativas más marcadas que ésta. Se absorbe bien por vía oral,
alcanzando niveles plasmáticos máximos en 0,5-2 horas (tabla 38-4). Se une en
el 70 % a la a1-glucoproteína ácida y se biotransforma en el 50 % a nivel
hepático, formándose metabolitos mono desalquilados activos. Su eliminación se
realiza fundamentalmente por vía renal, por lo que en ancianos y con
insuficiencia cardíaca o renal su semivida se prolonga desde 6-8 hasta 20
horas, acumulándose sus metabolitos.
La
disopiramida no modifica la digoxinemia. Fenitoína y rifampicina aceleran la
biotransformación y disminuyen los niveles plasmáticos de disopiramida. Las
reacciones adversas son similares a las descritas para la quinidina, aunque
produce más efectos anticolinérgicos e inotrópicos negativos que ésta. Las
acciones anticolinérgicas son más frecuentes en ancianos y contraindican su uso
en pacientes con glaucoma, retención urinaria, hipertrofia prostática,
miastenia grave o estreñimiento; estas reacciones se potencian por los
antidepresivos tricíclicos y pueden bloquearse con piridostigmina (90-180 mg
cada 8 horas). Deprime la contractilidad cardíaca y presenta propiedades
vasoconstrictoras que aumentan la poscarga y potencian la disminución del
volumen minuto, lo que contraindica su administración en arritmias asociadas a
insuficiencia cardíaca y su asociación con otros antiarrítmicos que también
deprimen la contractilidad (b-bloqueantes, verapamilo y diltiazem). En
pacientes con depresión de la contractilidad ventricular, la disopiramida puede
asociarse con digoxina, que no sólo aumenta la contractilidad, sino que
previene el aumento de la conducción AV que la disopiramida puede producir por
su potente acción antimuscarínica. La disopiramida aumenta las resistencias
vasculares periféricas y la presión arterial, pero su efecto inotrópico negativo
reduce el volumen minuto, por lo que la presión arterial apenas se modifica.
Está contraindicada si existe bradicardia, bloqueos AV e intraventriculares (en
pacientes sin marcapaso), shock, insuficiencia cardíaca, cuando aumenta el
intervalo QT del ECG y en las embarazadas, ya que estimula las contracciones
uterinas. Es efectiva en el tratamiento de las arritmias supraventriculares y
ventriculares no digitálicas (tablas 38-3 y 38-5) o asociadas al prolapso
mitral, aunque en el momento actual sólo se utiliza en pacientes en los que
otros fármacos han fracasado o se toleran mal. En arritmias supraventriculares
debe asociarse a digoxina para controlar sus efectos antimuscarínicos sobre el
nodo AV.
Por
su efecto inotrópico negativo es útil en el tratamiento de la miocardiopatía
hipertrófica obstructiva.

FÁRMACOS
ANTIARRÍTMICOS DEL GRUPO IB 1.
LIDOCAINA
Es
un anestésico local del tipo amida que presenta una alta afinidad por el estado
inactivo del canal de Na+ , siendo en la actualidad el fármaco de elección en
el tratamiento de las taquiarritmias ventriculares graves en medio
hospitalario.

PROPIEDADES
FARMACOLOGICAS
A
concentraciones terapéuticas no modifica la frecuencia sinusal, aunque la
disminuye en enfermos con disfunción sinusal previa, pero suprime el
automatismo del sistema de His-Purkinje, el automatismo anormal y la actividad
desencadenada por pospotenciales tempranos (acorta la duración del potencial de
acción) y tardíos.
Tampoco
altera la excitabilidad, la velocidad de conducción o el período refractario de
las células auriculares, del nodo AV (no altera el intervalo PR) o del
ventrículo sano (no altera el QRS del ECG). En el ventrículo isquémico,
parcialmente despolarizado (potencial de reposo ~70 mV), predomina el estado
inactivo del canal de Na+ , por el cual la lidocaína presenta mayor afinidad.
Como consecuencia, deprime aún más la excitabilidad y la velocidad de
conducción, pudiendo convertir las áreas de bloqueo unidireccional en
bidireccional y suprimir las arritmias por reentrada a concentraciones a las
que apenas modifica el miocardio sano circundante. A nivel ventricular, la
lidocaína acorta la duración del potencial de acción (los intervalos QT y JT) y
del período refractario ventricular, pero este acortamiento es más marcado en
las fibras de Purkinje que presentan un potencial de acción más prolongado que
en las fibras musculares ventriculares y en el miocardio isquémico que
presentan un potencial de acción más corto. El resultado es una repolarización
ventricular más homogénea que impide la reentrada del impulso cardíaco. A dosis
terapéuticas, los antiarrítmicos del grupo IB apenas modifican la presión
arterial, el volumen minuto o la contractilidad cardíaca, siendo de elección en
enfermos con insuficiencia cardíaca. Sin embargo, a dosis altas, pueden
producir hipotensión, bradicardia y depresión de la contractilidad y del
volumen minuto.
FARMACOCINETICA
La lidocaína sufre un intenso efecto de primer
paso hepático, por lo que la vía de elección es la IV, alcanzándose
concentraciones plasmáticas eficaces en 5 min. Se une en el 50 % a la
a1-glucoproteína ácida, distribuyéndose rápidamente por el organismo. En
pacientes con infarto de miocardio aumenta la fracción de lidocaína unida a
proteínas plasmáticas, lo que explica por qué algunos de ellos precisan dosis
muy altas del fármaco. Se biotransforma rápidamente en el hígado en
monoetilglicilxilidida (que presenta propiedades antiarrítmicas y
convulsivantes similares a las de la lidocaína) y glicinexilidida, que se
eliminan por vía renal. Esta rápida biotransformación y amplia distribución
explican por qué su semivida tras administración IV es de tan sólo 10 min.
En pacientes con insuficiencia cardíaca
disminuyen su distribución tisular y su biotransformación hepática, lo que
obliga a reducir la dosis del fármaco. Dado que la efectividad antiarrítmica de
la lidocaína depende de los niveles plasmáticos que alcanza en el
compartimiento central y por vía IV éstos disminuyen rápidamente (~8 min), se
administra en forma de bolo (50-100 mg) durante 15 min, seguida de una infusión
IV de 1-5 mg/min durante 24-48 horas. La velocidad de aclaramiento de la
lidocaína depende del flujo hepático, por lo que aquellas situaciones que
reducen el volumen minuto o el flujo sanguíneo hepático (insuficiencia
cardíaca, shock, cirrosis, b-bloqueantes, halotano o en ancianos) o la
actividad del sistema microsómico hepático (cimetidina y famotidina)
incrementan sus niveles plasmáticos, su distribución y su semivida (hasta
150-300 min en pacientes con insuficiencia cardíaca o hepática). En todas estas
situaciones se deberá reducir tanto la dosis del bolo inicial como la dosis de
mantenimiento. Por el contrario, la isoprenalina, que aumenta el flujo
sanguíneo hepático, y los inductores enzimáticos (fenitoína y rifampicina)
acortan su semivida.
REACCIONES
ADVERSAS
a)
neurológico (vértigo, euforia, disartria, nerviosismo, parestesias, temblor,
visión borrosa, diplopía, nistagmo, ataxia, confusión mental, depresión 38.
Fármacos antiarrítmicos 661 respiratoria y, a grandes dosis, convulsiones que
ceden con diazepam).
b) digestivo (náuseas y vómitos).
c) cardiovascular (depresión de la
contractilidad, bradicardia, bloqueo AV, hipotensión y ensanchamiento del QRS
con riesgo de aparición de efectos arritmogénicos).
En
cualquier caso, la lidocaína es el antiarrítmico que menor cardiode presión
produce. Su consumo está contraindicado en pacientes con historia previa de
alergia a los anestésicos locales de tipo amida, epilepsia, hepatopatías
graves, bradicardia, hipotensión y con bloqueo AV avanzado en pacientes sin
marcapaso, ya que al suprimir los ritmos de escape idioventriculares puede
producir un paro cardíaco.
Posee propiedades muy similares a las de la
lidocaína, presentando sobre ésta la ventaja de que puede administrarse por vía
oral.
Se absorbe bien por vía oral, alcanzando
niveles plasmáticos máximos por esta vía al cabo de 2 horas (tabla 38-4). Por
vía IV, sus efectos aparecen al cabo de 2-3 min. Se une en el 80 % a proteínas
plasmáticas y se distribuye ampliamente atravesando la barrera
hematoencefálica. El 90 % de la dosis se biotransforma a nivel hepático,
retrasándose su absorción y biotransformación en pacientes con insuficiencia
hepática o cardíaca, infarto de miocardio o que reciben analgésicos narcóticos.
Se elimina lentamente por vía renal (semivida: 8-12 horas), aumentando sus
niveles plasmáticos y su semivida en hepatopatías y postinfarto de miocardio
(18 horas), pero no si existen nefropatías. El aclaramiento renal de la
mexiletina aumenta al acidificar la orina. Fenitoína y rifampicina aceleran la
biotransformación de la mexiletina y disminuyen sus niveles plasmáticos,
mientras que la mexiletina aumenta los niveles plasmáticos de teofilina.

REACCIONES
ADVERSAS
Produce reacciones adversas digestivas
(anorexia, náuseas, vómitos y dispepsia en el 20 % de los pacientes),
neurológicas y cardiovasculares similares a las de la lidocaína. Su uso está
contraindicado si hay hepatopatías graves, bradicardia, hipotensión y bloqueo
AV avanzado en pacientes sin marcapaso ya que, al suprimir los ritmos de escape
idioventriculares, puede producir un paro cardíaco.
USOS
E INDICACIONES
Se
utiliza en la profilaxis y tratamiento de taquiarritmias ventriculares
postinfarto de miocardio. El tratamiento puede iniciarse con una dosis de
sobrecarga por vía oral (400 mg seguidos al cabo de 2-6 horas de 300-1.200 mg)
o IV (bolo de 100-250 mg, seguidos de 2 mg/kg/h durante 3,5 horas y,
finalmente, por una dosis de mantenimiento de 0,5- 1 mg/min).
APRINDINA
Es una amina terciaria que, a diferencia de la
lidocaína y la mexiletina, reduce la frecuencia sinusal, deprime la
excitabilidad y la velocidad de conducción intraventricular, la contractilidad
cardíaca y prolonga los períodos refractarios cardíacos.
Se
absorbe bien por vía oral, llevando a cabo su acción al cabo de 2 horas (tabla
38-4), aunque su efectividad máxima se observa al cabo de varios días de
tratamiento. Por vía IV su acción aparece a los 5- 10 min. Se une en el 95 % a
proteínas plasmáticas y tisulares. Sufre glucoconjugación hepática,
convirtiéndose en N-desetilaprindina, que posee también actividad
antiarrítmica, eliminándose ambas por orina (70 %) y heces (30 %).
Su
prolongada semivida (unas 30 horas) permite una cómoda dosificación por vía
oral. La aprindina posee un estrecho margen terapéutico produciendo reacciones
digestivas (náuseas, vómitos y diarrea), neurológicas (nerviosismo, temblor,
ataxia, vértigo, somnolencia, fatiga, diplopía, convulsiones y alucinaciones) y
cardiovasculares (depresión de la función ventricular, hipotensión, bloqueos AV
e intraventriculares y ensanchamiento del QRS).
REACCIONES
ADVERSAS
Todas
estas reacciones son más frecuentes al comienzo del tratamiento y en particular
si se utilizan dosis de choque. Ictericia colestásica y agranulocitosis son
reacciones idiosincrásicas raras, no relacionadas con la dosis y que pueden
revertirse tras la supresión del fármaco. La aprindina está contraindicada en
embarazadas.
Por
su estrecho margen terapéutico, se utiliza en la profilaxis y tratamiento de
arritmias ventriculares, síndrome de Wolff-Parkinson White (WPW) o taquicardias
supraventriculares paroxísticas refractarias a otros tratamientos.
FÁRMACOS ANTIARRÍTMICOS DEL GRUPO IC
Presentan una elevada afinidad por el estado
activo del canal de Na+ y prolongan marcadamente su reactivación, siendo los
fármacos del grupo I que más deprimen la INa y, por lo tanto, los que mayor
depresión de la excitabilidad y conducción intracardíaca producen, así como
mayor incidencia de efectos arritmogénicos.
PROPAFENONA
Este fármaco no sólo inhibe la INa (grupo I),
sino que también es capaz de inhibir diversas corrientes de salida de K+ (grupo
III), la ICa (grupo IV) y bloquea los receptores b1-adrenérgicos cardíacos
(grupo II).

ACCIONES
FARMACOLOGICAS
Por
sus acciones inhibidoras de la INa e ICa y b-bloqueantes, la propafenona
disminuye la frecuencia sinusal y suprime el automatismo del sistema de
His-Purkinje, el automatismo anormal y la actividad desencadenada por
pospotenciales tempranos o tardíos. A concentraciones terapéuticas, la
propafenona inhibe la INa y deprime la excitabilidad y la velocidad de
conducción intraauricular e intraventricular, siendo su acción más marcada a la
altura del sistema de His-Purkinje (ensancha el QRS). A nivel ventricular,
acorta la duración del potencial de acción y del período refractario de las
fibras de Purkinje, pero si apenas altera estos parámetros en las fibras
musculares ventriculares, el resultado es una mayor disparidad en la
recuperación de la excitabilidad entre fibras adyacentes que facilitaría la
aparición de arritmias por reentrada. Estos efectos contrapuestos sobre la
duración del potencial de acción explican por qué apenas si modifica el
intervalo JT del ECG, por lo que la prolongación del intervalo QTc sería
consecuencia de la marcada prolongación del complejo QRS que estos fármacos
producen. También deprime la velocidad de conducción (prolonga el intervalo PR)
y prolonga el período refractario del nodo AV y de las vías accesorias AV,
siendo útil para controlar la frecuencia ven662 Farmacología humana tricular si
existen taquicardias supraventriculares y de la unión AV. A dosis altas, la
inhibición de la INa puede llegar a bloquear la conducción del impulso
cardíaco, facilitándose la reentrada del impulso cardíaco (v. más adelante).
Este efecto, unido a la mayor disparidad de la recuperación de la excitabilidad
ventricular explica por qué los antiarrítmicos del grupo IC son los que mayor
incidencia de efectos arritmogénicos presentan. En pacientes con función
ventricular normal, su administración oral apenas modifica la contractilidad
ventricular, las resistencias vasculares periféricas, la presión arterial o el volumen
minuto. Sin embargo, por vía IV rápida o en pacientes con insuficiencia
cardíaca previa, producen una clara depresión de la contractilidad y de la
fracción de eyección ventriculares, acentuando o desencadenando un cuadro de
insuficiencia cardíaca.
FARMACOCINETICA
Se absorbe bien por vía oral, pero su
biodisponibilidad es baja y dosis-dependiente (5-30 %), lo que indica que sufre
un importante efecto de primer paso hepático (tabla 38-4). Su acción aparece al
cabo de 30 min, alcanza su efecto máximo al cabo de 2-4 horas y se prolonga
durante 8 horas. Por vía IV, su efecto es inmediato y se prolonga durante 2
horas. Se une en el 95 % a la a1-glucoproteína ácida y se distribuye
ampliamente, biotransformándose en el 99 % en diversos metabolitos, de los que
la 5-hidroxipropafenona es más potente que la propia propafenona para inhibir
la INa; éste y otros metabolitos se eliminan conjugados por vía renal. La
semivida aumenta durante el tratamiento, lo que sugiere que el sistema de
biotransformación hepática se satura y existen importantes diferencias
individuales en la velocidad de su biotransformación por el citocromo P-450
2D6. En los metabolizadores rápidos (90 % de la población) su semivida es de
5-6 horas y en los lentos de 15-17 horas; estas diferencias explican la pobre
relación existente entre la dosis y la respuesta terapéutica.
REACCIONES
ADVERSAS
Durante
el tratamiento aparecen reacciones digestivas (anorexia, náuseas, sensación de
plenitud, sabor metálico e ictericia colestásica) y neurológicas (mareos,
temblor, nerviosismo, inestabilidad motora, parestesias y visión borrosa),
aunque las más graves son las cardiovasculares, caracterizadas por depresión de
la contractilidad, hipotensión, bradicardia, bloqueos AV e intraventriculares y
taquiarritmias ventriculares. Sus acciones proarrítmicas aparecen en el 5-15 %
de los pacientes y son más frecuentes en aquéllos con insuficiencia cardíaca,
síndrome de QT largo, trastornos previos de la conducción intraventricular,
isquemia o cardiopatías estructurales, es decir, en aquellos pacientes que más
necesitan el tratamiento antiarrítmico. La propafenona está contraindicada en
pacientes con hipotensión, shock cardiogénico, enfermedad del seno,
bradicardia, insuficiencia cardíaca o bloqueos AV o intraventriculares (a menos
que se haya implantado un marcapaso para mantener la frecuencia ventricular).
La propafenona, por su acción b-bloqueante, podría estar contraindicada en
metabolizadores lentos asmáticos. La asociación de propafenona con b-bloqueantes,
disopiramida, verapamilo o diltiazem aumenta la incidencia de bradicardia,
bloqueo AV e insuficiencia cardíaca. Su asociación con antiarrítmicos del grupo
IA aumenta la incidencia de arritmias cardíacas y, con digoxina, la incidencia
de bradicardia y bloqueo AV. La propafenona aumenta los niveles plasmáticos de
anticoagulantes orales, digoxina y metoprolol, mientras que la cimetidina
aumenta los de propafenona.
FLECAINIDA
Es
un derivado benzatínico que presenta propiedades electrofisiológicas similares
a las de la propafenona, aunque a diferencia de ésta, carece de propiedades
b-bloqueantes. Se emplea por vía oral, alcanzando su efecto máximo al cabo de
90 min. Se une a proteínas plasmáticas en el 37-48 % y se biotransforma en el
75 % en el hígado, formándose metabolitos conjugados inactivos que se eliminan
junto con el fármaco no biotransformado por vía renal. La eliminación renal
disminuye al alcalinizar la orina. Su semivida es de unas 13 horas y aumenta
hasta 14-26 horas en pacientes con insuficiencia cardíaca, arritmias
ventriculares o insuficiencia renal.
Durante
el tratamiento aparecen reacciones digestivas, neurológicas y cardiovasculares
similares a las producidas por la propafenona. La flecainida, además, produce
ictericia colestásica y visión borrosa. La flecainida también está
contraindicada en pacientes con hipotensión, shock cardiogénico, enfermedad del
seno, bradicardia, insuficiencia cardíaca o bloqueos AV o intraventriculares (a
menos que se haya implantado un marcapasos para mantener la frecuencia
ventricular).
La
asociación de flecainida con b-bloqueantes, disopiramida, verapamilo o
diltiazem aumenta la incidencia de bradicardia, bloqueo AV e insuficiencia
cardíaca y su asociación con antiarrítmicos del grupo IA aumenta la incidencia
de arritmias cardíacas y, con digoxina, la incidencia de bradicardia y bloqueo
AV.
La cimetidina aumenta los niveles plasmáticos
de flecainida y ésta, al mismo tiempo, los de digoxina y propanolol, a la vez
que potencia el efecto inotrópico negativo de éste. La amiodarona aumenta los
niveles plasmáticos de flecainida, debiendo reducirse la dosis de ésta a la
mitad si ambos fármacos se asocian.


RELACIONADAS
CON EL PACIENTE
Preguntarles a
estos pacientes si han tenido tratamiento previo con digitálicos y precisar
dosis, vía de administración, frecuencia diaria, tiempo de duración, si fue
necesario suspender el tratamiento y su causa.
Resulta
imprescindible conocer el peso corporal para realizar el cálculo de la dosis
corporal total del digitálico que se va a administrar, la cual estará
relacionada con el funcionamiento de los sistemas renal, hepático, tiroideo,
pulmonar y cardiaco, determinado a través de la anamnesis, el examen físico y
los exámenes complementarios que, de estar alterados, provocarían una disminución
del aclaramiento plasmático, igual que sucede con algunas enfermedades, tales
como amiloidosis, enfermedad pulmonar obstructiva crónica (EPOC), anemias,
valvulopatías, miocardiopatía hipertrófica (en el hipotiroidismo la vida media
de la digital está aumentada y en el hipertiroidismo, disminuida). Dichos
elementos, además de la edad avanzada, están estrechamente relacionados con una
enfermedad cardiaca grave que necesitará un tratamiento óptimo para evitar la
sobredosificación y finalmente la intoxicación digitálica. Asimismo, otro
elemento favorecedor de la toxicidad es el desequilibrio electrolítico
(hipopotasemia, hipomagnesemia o hipercalcemia).
Durante el tratamiento con digitálicos es
necesario realizar cada 6 meses un electrocardiograma (ECG) y pruebas sobre las
funciones renal y hepática para detectar precozmente signos
electrocardiográficos de impregnación digitálica o un deterioro progresivo de
la función de estos órganos, siendo factores de riesgos para la intoxicación digitálica;
por tanto, si se manifestara alguna alteración habría que reajustar o suspender
la dosis de estos digitálicos.
MECANISMO DE ACCION
El efecto
inotrópico positivo (leve o moderado) es consecuencia de la inhibición de la
ATPasa de la membrana celular y no depende de la acción inotrópica positiva de
las catecolaminas, por ende, existe respuesta inotrópica positiva en presencia
de bloqueo beta adrenérgico debido a que se bloquea la bomba de sodio, lo cual
origina un aumento en la concentración intracelular de sodio y calcio que
favorece la contracción del músculo liso cardiaco. Esta acción de estimular la
contractilidad miocárdica está ligada a una rapidez en la evolución del
síndrome de insuficiencia cardiaca; además, la respuesta inotrópica positiva
aumenta de manera lineal hasta la toxicidad e incrementa el riesgo de muerte,
pero también conlleva a beneficios, tales como mejoría de la sintomatología del
paciente y en su calidad de vida, disminución de la frecuencia de ingreso, de
la fracción de eyección y la capacidad de realizar esfuerzo sobre todo cuando
se asocia a diuréticos e inhibidores de la enzima convertidora de la
angiotensina II (IECA) especialmente en los pacientes con cardiomegalia y en
los que presentan los grados III-IV de la clasificación de la Asociación del
Corazón de Nueva York (NYHA).
Los digitálicos potencian la acción vagal
sobre el corazón y disminuyen el grado de activación neurohormonal; además,
alargan el periodo refractario funcional de la unión auriculoventricular (AV) y
disminuyen la respuesta ventricular, por este efecto son útiles en el
tratamiento de pacientes con fibrilación y aleteo auricular, mientras que la
disminución del potencial de reposo y el aumento de la excitabilidad del
miocardio explica las arritmias producidas por toxicidad digitálica; (12,16)
sin embargo, también se plantea que los digitálicos pueden producir un efecto
vasoconstrictor en la circulación normal y quizás en el paciente con
insuficiencia cardiaca. Los estudios han demostrado que este efecto está
relacionado con la rápida administración de los digitálicos por vía
intravenosa, pero no existe evidencia de que este efecto se manifieste con la
dosis oral.
El mecanismo de la toxicidad es estrictamente
celular y se explica de la manera que sigue:
- La sobrecarga de calcio intracelular retrasa
la despolarización dependiente de calcio al aumentar el potencial de membrana
en reposo y hace la célula más excitable, lo cual conduce al automatismo
ventricular.
- Exagerada estimulación vagal [bradicardia y
bloqueo auriculoventricular (BAV)]
- Efecto
depresor directo de la digoxina sobre el nodo auriculoventricular
- Todo lo anterior conlleva a la disfunción
miocárdica.
INDICACIONES
ACTUALES SOBRE LOS DIGITALICOS
- Para tratar
pacientes con arritmias supraventriculares (específicamente aleteo, fibrilación
auricular (FA) con respuesta ventricular acelerada y taquicardia paroxística
supraventricular TPSV) en dosis mayores que para los afectados con
insuficiencia cardiaca; serán tóxicos en otros eventos cardiacos.
- Insuficiencia
ventricular izquierda aguda, tipo edema agudo de pulmón (EAP) que no tenga de
base un infarto agudo de miocardio (IMA).
- Insuficiencia ventricular izquierda aguda
con disfunción sistólica y fibrilación auricular.
- Insuficiencia
cardiaca crónica con disfunción sistólica, cardiomegalia y tercer ruido.
- Pacientes con
fracción de eyección deprimida (FE menor de 40 %) que lleven tratamiento con
betabloqueadores (BB) para el control de la frecuencia cardiaca a largo plazo.
- Afectados
incluidos en la clase III-IV de la NYHA con disfunción ventricular sistólica
sintomática en ritmo sinusal o FA que reciban tratamiento combinado con
betabloquedores, inhibidores de la enzima corvertidora de la angiotensina I en
angiotensina II (IECA) y antialdosterónicos.
- Cor- pulmonale crónico (CPC) severo y
refractario al tratamiento.
CONTRAINDICACIONES
- En pacientes
diagnosticados con FA y respuesta ventricular acelerada en el curso de un
síndrome de preexcitación de Wolff Parkinson White (WPW), pues favorece la
perpetuación de la arritmia al bloquear la vía fisiológica y dejar intacta la
vía accesoria, que son conexiones anormales que permiten la conducción del
impulso entre las aurícula y los ventrículos a través del anillo fibroso
auriculoventricular (AV); estas aparecen desde el nacimiento debido a la
participación incompleta de las aurículas y los ventrículos por parte de los
anillos fibrosos AV, con diferentes localizaciones a través de estos y del
tabique, muy a menudo entre la aurícula izquierda y la pared libre del
ventrículo izquierdo, seguido de otros sitios posteroseptal, pared libre
derecha, y anteroseptal; por la cual se conduce el impulso desde la aurícula
hacia el ventrículo (conducción anterógrada) y origina la taquiarritmia con
todas sus características.
- La insuficiencia renal es una
contraindicación relativa.
- Grados
avanzados de BAV (segundo o tercer grado) y bloqueo cardiaco intermitente.
- Pacientes con
EPOC, corpulmonale crónico clasificado en leve o moderado por el grado de
hipoxia mantenido, la cual favorece la intoxicación digitálica a través de la
alteración del balance hidromineral, puesto que el descenso en la
disponibilidad de oxigeno (O2) para las células inhibe la fosforilación
oxidativa y aumenta la glucólisis anaeróbica, conocido como efecto Pasteur, así
como mantiene reducida la producción de 5- trifosfato de adenosina (ATP) que al
volverse insuficiente para garantizar el equilibrio iónico y osmótico provoca
la despolarización de la membrana celular y permite la entrada descontrolada de
iones calcio (ca2+), lo cual provoca la contracción del músculo liso y coincide
con el mecanismo de acción de los digitálicos que depende de los iones
(Na+yCa2+); además, la hipoxia favorece la activación de fosfolipasas proteasas
que producen edema y muerte celular. Otro electrolito que participa activamente
es el ión potasio K+ porque la inhibición de sus conductos causa
despolarización y activa los conductos del ca2+ e induce la contracción de la
célula de la musculatura lisa. Durante la hipoxia se estimula el arco reflejo
de los quimiorreceptores para inducir venoconstricción y la vasodilatación
acompañada del incremento de la contractilidad miocárdica transitoria en la
hipoxia aguda, mientras que en la crónica aparece el efecto contrario que es la
disminución de la contractilidad miocárdica.
- Afectados con diagnóstico de miocardiopatía
hipertrófica, puesto que aumenta el grado de obstrucción del tracto de salida
del ventrículo izquierdo (VI).
- Individuos diagnosticados con infarto agudo
del miocardio, pues aumenta el consumo de oxigeno del corazón y favorece su
expansión; además el remodelado ventricular izquierdo que son los cambios
ocurridos en el corazón en forma, tamaño, estructura celular y funcionamiento
(dilatación, balonamiento e hipertrofia ventricular) como consecuencia de la isquemia
y de la hipertensión arterial; los digitálicos son medicamentos arritmogénicos,
lo cual puede conllevar a la ruptura cardiaca.
- Sospecha de
intoxicación digitálica.
- Fibrilación
ventricular.
- Alteraciones electrolíticas, tales como hipopotasemia,
hipomagnesemia
DIGITALIZACION
Para el tratamiento agudo (FA, aleteo o flúter
auricular y EAP) la dosis a emplear es de 1- 1,5 mg en 24 horas por vía
intravenosa (IV), que siempre será inferior a la vía oral debido a que su
absorción es incompleta por esta última vía; la intramuscular es inconsistente
y extremadamente dolorosa, se presenta en ámpula de 0,25 mg/2 mL. Teniendo en
cuenta las características de los pacientes a los cuales se les va a indicar
(edad avanzada) y como se conoce la eficacia de la digoxina en el control de la
respuesta ventricular en el reposo, se utilizan dosis de 0,5-1 mg por 24 horas;
se comienza con 0,25 mg por vía intravenosa, repetir a los 30 minutos de ser
necesario y de ahí cada 4 u 8 horas 0,25 mg hasta completar 1 mg en 24 horas o
emplear 0,25 mg cada 6 hasta 24 horas. Los digitálicos tienen acción rápida
(5-10 minutos).
Para la
administración por vía oral (tableta de 0,25 mg) se debe calcular el contenido
orgánico total deseable (mg/Kg), digoxina 0,01-0,02 mg/kg; el objetivo es
sustituir las pérdidas diarias del fármaco que oscilan entre 37 % del total con
un funcionamiento renal normal y 14 % en la insuficiencia renal terminal. La
dosis calculada se puede administrar de forma única sino es muy elevada, con
alto riesgo de toxicidad; lo ideal es indicarla en un plazo de 24-72 horas y
luego la dosis de mantenimiento que corresponde según el funcionamiento renal
del paciente.
En la actualidad se prefiere emplear los
digitálicos lentamente para evitar el riesgo de toxicidad. Se debe comenzar con
dosis de mantenimiento de 0,25 mg/día en aquellos pacientes con función renal
normal; para garantizar mayor protección se recomienda descansar sábados y
domingos, en cualquier esquema que se utilice independientemente de la función
renal.
En caso de que
esta última sea insuficiente o se trate de un paciente con edad muy avanzada
los autores, según su experiencia, utilizan 0,125 mg/día o 0,25 mg en días
alternos con descanso el fin de semana, es decir, la insuficiencia renal no
contraindica el uso de la digoxina mientras que se administre teniendo en
cuenta el filtrado glomerular (lo que el riñón pueda excretar). Cabe decir que
en los digitálicos las dosis óptima y tóxica no varían sensiblemente de un
preparado a otro; la dosis letal es 5-10 veces superior a la mínima efectiva.
El cuadro clínico de la toxicidad se expresa con cifras en sangre superiores a
3-4 veces su valor normal. Los síntomas cardiovasculares se presentan con
niveles por encima de 5ng/mL y la parada cardiorrespiratoria con 10ng/mL, pero
cuando el tenor en sangre sea de 2ng/mL o más debe constituir un alerta para el
médico, quien revaluará la dosificación; por ello es importante que
periódicamente se realice la determinación sérica de los digitálicos en
pacientes que llevan tratamiento prolongado con estos fármacos o tienen algún
factor de riesgo favorecedor de la toxicidad.(10) La concentración terapéutica
es de 1-1,5ng/mL. Hasta hace una década se tomaba como referencia 2 ng/mL, pero
debido a que el índice terapéutico es muy cercano al nivel tóxico se pretende
que el afectado mantenga su nivel menor a 1ng/mL sobre todo si tiene una edad
avanzada, poca masa corporal y alteración de la función renal.
INTERRACION MEDICAMENTOSA
Entre los
fármacos de uso frecuente en el tratamiento de cardiopatías y arritmias que
aumentan la vida media de la digital y elevan su nivel sérico en 70-100 %
figuran: amiodarona, quinidina, procainamida, espironolactona, verapamilo,
propafenona y flecainide. Existen otros, tales como eritromicina, tetraciclina
y omeprazol que aumentan su absorción y consecuentemente los niveles séricos en
40-100 %, entonces se deben indicar con precaución, puesto que favorecen la intoxicación
digitálica. (10,25) Cabe agregar que el empleo combinado de digitálicos con
fármacos como la metildopa, los betabloqueadores, bloqueadores de los canales
del calcio y la clonidina pueden originar bloqueos auriculoventriculares y
bradicardia sinusal al prolongar el retraso fisiológico de la conducción del
estímulo a nivel del nodo A-V.
RELACIONADA CON
EL TRATAMIENTO DIGITALICO
Los pacientes
con riesgo de toxicidad digitálica deben ser tratados con dosis pequeñas y a
todos se les debe medir la concentración sanguínea de digital. (27) En este
medio, la respuesta clínica es el parámetro para definir la cantidad de fármaco
a administrar, debido a la dificultad en la determinación sanguínea de dichos
pacientes. No deben retirarse los digitálicos en los individuos que ya los
reciban si la indicación ha sido acertada, puesto que se ha demostrado que
estos individuos revierten a los signos de insuficiencia una vez que se
descontinúa la digoxina. Las dosis masivas de digital producen hipercalcemia al
producir vasoconstricción mesentérica y coronaria, lo que origina isquemia o
infarto mesentérico; en caso de intoxicación o sobredosis se recomienda emplear
el anticuerpo antidigoxina.
Los signos de impregnación digitálica se
manifiestan por la denominada cubeta digitálica que está constituida por una
disminución del segmento ST de concavidad superior y acortamiento de intervalo
QT.
Inmunoterapia antidigoxina (fragmento de
anticuerpo fab)
La inmunoterapia antidigoxina es la aplicación
del anticuerpo contra la digoxina utilizado en el tratamiento de pacientes con
intoxicación digitálica aguda o crónica, obtenido a partir del antisuero de
digoxina específico, el cual es capaz de revertir la intoxicación grave.
MECANISMO DE
ACCION
Se une a la
digoxina libre en el suero para inactivarla y así evitar la unión a los
receptores, debido a esto los niveles de digitálicos en sangre se incrementarán
después de la terapia; por tal razón, no deben considerarse de primera línea en
el tratamiento. Su excreción es por vía renal, su efecto empieza en pocos
minutos y se logra la reversión completa en 30 minutos.
EFECTOS
ADVERSOS
Reacciones de
anafilaxia, insuficiencia cardiaca por la abolición del efecto terapéutico de
la digoxina, enfermedad del suero, hipopotasemia e intoxicación de rebote en
pacientes con insuficiencia renal causado por la digoxina libre remanente,
debido a su mecanismo de acción; el control rápido de la hiperpotasemia con
fragmentos de anticuerpo antidigoxina puede conducir en las primeras 4 horas a
una hipopotasemia, que refleja la eficacia del tratamiento y ocasionalmente
requiere de la administración del cloruro potásico.
INDICACIONES PRECISAS EN LA INTOXICACION AGUDA Y CRONICA
Alteraciones
que amenazan directamente la vida, tales como taquicardia o fibrilación
ventriculares y bradiarritmias, que incluye la bradicardia sinusal sintomática
con frecuencia cardiaca por debajo de 40 pulsaciones por minuto y los bloqueos
auriculoventriculares de segundo y tercer grados, ambas refractarias a la
atropina. También se indica en la asistolia ventricular y en la hiperpotasemia
superior a 5 meq/L, digoxinemia de 10 ng/mL o más en la intoxicación aguda;
además, en pacientes que han ingerido más de 10 mg de digoxina que es
equivalente a 40 comprimidos de 0,25 mg y en aquellos que presenten síntomas y
signos rápidamente progresivos de toxicidad digital, así como choque
cardiogénico.
Intoxicaciones potencialmente letales como la
bradicardia ligera con frecuencia cardiaca menor de 60 pulsaciones por minuto
en adultos con factores de pronóstico negativo asociado, sin tener en cuenta
problemas de conducción. En estos pacientes, cuando la atropina no eleva las
pulsaciones por encima de 60, se administra la mitad de una dosis equimolar o
profiláctica y se monitoriza, puesto que en todos los casos no es necesaria una
dosis completa de anticuerpo antidigoxina, y en la intoxicación crónica la
dosis es de 6 bulbos o vial de 40 mg.
DOSIFICACION
La dosis de anticuerpo en la intoxicación
digitálica está determinada por el peso corporal del paciente, los miligramos
de digital ingeridos y los niveles séricos de este. Luego se procede a aplicar
las siguientes fórmulas:
- Cuando se
conocen los miligramos administrados N0 viales = Dosis ingeridas en mg × 0,8 ÷
0,5
- Cuando se conoce la digoxinemia N0 viales =
Digoxina sérica en ng/mL × kg de peso corporal ÷ 100
Si se desconoce la dosis del medicamento que
se ha empleado o su concentración sérica se debe administrar 20 vial de 40 mg
en infusión intermitente durante 15 a 40 minutos, o directa en vena en caso de
parada cardiorespiratoria, cada vial o bulbo neutraliza a 0,5 mg de digital. Se
ha sugerido administrar dicha dosis en 2 partes iguales, la primera mitad en
unos 15 a 30 minutos y la segunda, en unas 7 horas, con el objetivo de
disminuir el riesgo de recurrencia de la intoxicación.
PREPARACION
Reconstituir
cada vial de 40 mg con 4 mL de agua estéril, luego diluir la dosis a
administrar en 50-100 mL de solución salina al 0,9 %; una vez preparado el
medicamento puede conservarse en una nevera de 2- 80 por 4 horas y protegerlo
de la luz. Se administra en forma directa o intermitente según sean las
condiciones del paciente
CLINICA
Se considera
que la intoxicación digitálica es una entidad frecuente, pero por sus síntomas
larvados e imprecisos iniciales, entre ellos los digestivos, puede pasar
inadvertida para el médico sobre todo en intoxicaciones crónicas, si no se correlacionan
los datos clínicos con los hallazgos electrocardiográficos, que son más
sugestivos. Todos estos síntomas son más evidentes en los ancianos con
tratamiento prolongado.
- Gastrointestinales: anorexia, náusea,
vómitos, malestar abdominal, hemorragia gastrointestinal y necrosis esofágica,
gástrica o intestinal.
- Neuropsiquiatría: percepción ocular de color
verde-rojizo, visión borrosa, de candelilla, diplopía, ambliopía, fotofobia,
alucinaciones, pesadillas, afasia, neuralgia del trigémino, parestesia,
amnesia, depresión, somnolencia, confusión y delirio.
- Arritmias cardiacas: son a menudo la
expresión inicial de cardiotoxicidad de la sobredosificación digitálica.
Se han descrito
casi todos los tipos de arritmias, y cualquier nuevo cambio del ritmo que
aparezca después de su administración debe ser considerado como sospechoso, las
más frecuentes son bradicardia sinusal, fibrilación auricular con respuesta
ventricular menor de 50 latidos por minuto, extrasístoles ventriculares en
forma de bigeminismo y la taquicardia bidireccional aunque se presentan otras,
tales como se muestra a continuación:
- Extrasístoles ventriculares bigeminados y trigéminas
- Bradicardia y
paro sinusales
- Bloqueos
sinoauricular y auriculoventricular: bloqueos de segundo grado, tipo Wenckebach,
bloqueos completos y con taquicardia auricular paroxística asociada.
- Taquicardia de la unión no paroxística y
taquicardia ventricular bidireccional
- Fibrilación auricular con respuesta
ventricular conservada o acelerada
- Ritmo
idioventricular acelerado

FARMACOS ANTIHIPERTENSIVOS
Diuréticos Los
diuréticos aumentan el volumen urinario alterando el manejo del sodio (Na+ ) a
nivel renal, resultando en la excreción de agua. A través de estos efectos en
el balance hidroelectrolítico, los diuréticos reducen el volumen sanguíneo y la
presión venosa, disminuyendo la precarga y el volumen de eyección cardíaco, lo
que lleva a la reducción de la PA.
Estos fármacos se clasifican en tres grupos:
● Tiazidas:
piedra angular del tratamiento hipertensivo desde hace décadas (ej.:
hidroclorotiazida (HCTZ), clortalidona (CTDN), indapamida).
● Diuréticos de
asa: potente efecto diurético y natriurético. Su uso está reservado para los
síndromes edematosos y sólo en raras ocasiones se utiliza por sus propiedades
antihipertensivas (ej.: furosemida). Debiese reservarse en casos de falla renal
o cardíaca.
● Ahorradores
de potasio (K+): por lo general no actúan sobre transportadores de Na+ lo que
les otorga una baja potencia diurética. Esta razón ha llevado a clasificar a
este grupo como un antagonista de la aldosterona en vez de un diurético, por lo
que su análisis se realizará en la sección respectiva.
INHIBIDORES ENZIMA COONVERTIDORA ANGIOTENSINA
Los IECA actúan
sobre el sistema renina-angiotensina-aldosterona (RAA). Dentro de este sistema
la angiotensina es una hormona peptídica que causa vasoconstricción y
adicionalmente estimula la liberación de aldosterona. Esta última promueve la
retención de Na+ a nivel renal, aumentando el volumen sanguíneo. Este grupo
farmacológico, tal como su nombre lo señala, inhibe a la enzima que convierte
la angiotensina-I en angiotensina-II, disminuyendo la síntesis de esta última y
bloqueando así el eje RAA.
A nivel internacional existen más de 11 IECA,
todos con similares indicaciones terapéuticas, efectos adversos y
contraindicaciones. Las diferencias se encuentran principalmente en dos
características: función directa o a través de un metabolito, y propiedades
farmacocinéticas. Prácticamente todos son de eliminación renal (24). Un metaanálisis
de Eran y cols. demostró que no hay diferencia significativa en la potencia
antihipertensiva entre los IECA disponibles, 60-70% de la magnitud de la baja
de PA se logra con las dosis iniciales recomendadas por el fabricante.
Adicionalmente, se ha demostrado que los IECA
son ideales para el paciente diabético ya que aumentan la sensibilidad a la
insulina, retrasan la nefropatía diabética y presentan un efecto antiproteinúrico.
El estudio MDRD (Modification of Diet and Renal Disease) concluyó que en
pacientes con enfermedad renal no asociada a diabetes, los IECA también son
nefroprotectores, especialmente en pacientes con proteinuria.
En personas con insuficiencia cardíaca con
altos niveles de angiotensina II, el tratamiento con IECA retrasa el inicio de
síntomas, previene mortalidad por falla cardíaca, y disminuye la frecuencia de
hospitalizaciones.
También se han
realizado extensos ensayos clínicos en personas post-IAM que tienen mayor
riesgo de presentar otro evento CV, entre los que se destacan los estudios SAVE
(Sleep Apnea Cardiovascular Endpoints Study) (28), AIRE (Acute Infarction
Ramipril Efficacy) (29) , TRACE (Trandolapril Cardiac Evaluation) (30) y SMILE (The
Survival of Myocardial Infarction Long-term Evaluation) (31). Ellos han
demostrado convincentemente que los IECA mejoran la sobrevivencia post-IAM,
especialmente cuando existe disfunción del ventrículo izquierdo o congestión
pulmonar aguda.
BLOQUEADORES DE CANALES DE CALCIO
Los BCC son
potentes antihipertensivos que actúan bloqueando los canales de calcio (Ca++)
voltajedependiente en el músculo liso vascular, miocitos del nodo
auriculoventricular y del nodo sinoauricular. El principal mecanismo de acción
es la disminución de la resistencia arterial periférica provocada por la
disminución de la entrada de Ca++ al intracelular. Además, han mostrado ser
eficaces en la regresión tanto de la hipertrofia ventricular izquierda, como de
la albuminuria moderada y el grosor de la placa de ateroma carotídeo.
Los BCC se
dividen en dos subclases, dihidropiridínicos (DHP) y no dihidropiridínicos
(NDHP). Los primeros tienen mayor selectividad vascular además de un efecto
natriurético y los segundos mayor selectividad miocárdica e inhibición del
sistema de conducción.
Los BCC DHP actúan principalmente a nivel del
músculo liso y se dividen en fármacos de acción corta, como el nifedipino, y de
acción prolongada, dentro de los cuales destaca el amlodipino, nifedipino
retard, nitrendipino y felodipino. Los BCC NDHP, verapamilo y diltiazem, actúan
sobre el sistema de conducción y miocardio contráctil, siendo además efectivos
en el tratamiento HTA asociado a arritmias.
El metaanálisis
de Chen y Yan que incluye a 273,543 personas concluyó que los BCC disminuyen la
tasa de ACV en pacientes hipertensos al compararlos con el uso de placebo.
Los BCC no producen alteraciones metabólicas
en la glicemia, perfil hepático, ni balance electrolítico, siendo un fármaco
seguro de usar en adultos mayores, personas con diabetes y en aquellos en que
los diuréticos están contraindicados.
El edema de
extremidades inferiores (perimaleolar) es la principal reacción adversa a estos
medicamentos y una causa frecuente de abandono. Es dosis dependiente, se presenta
usualmente luego de 3 meses de tratamiento, con una prevalencia del 26% a los 6
meses de inicio del tratamiento, y no responde al uso de diurético.
El nifedipino no debe ser empleado en el
tratamiento de la HTA crónica dado que tiene una vida media de 2-5 horas por lo
que requiere múltiples dosis al día, lo que se asocia a variaciones plasmáticas
significativas.
Tampoco debe ser utilizado en el tratamiento
de las crisis 22 hipertensivas ya que produce una caída brusca de la PA
acompañada de taquicardia refleja y activación del eje renina angiotensina,
aumentando el riesgo de isquemia miocárdica.
El amlodipino es el BCC de elección para el
tratamiento crónico de HTA dado que produce una disminución gradual de la PA y,
a diferencia de los otros BCC, tiene una vida media larga (40 h) lo que permite
su dosificación una vez al día, Tabla 4. Tiene una afinidad 10 veces mayor para
el tejido vascular periférico que para el tejido cardíaco.
Además,
presenta múltiples efectos pleiotrópicos actuando como anti ateroesclerótico
reduciendo el colesterol LDL, posee efectos antioxidantes, y favorece la
producción de óxido nítrico (53). Dado lo anterior es un buen fármaco en
adultos mayores. El amlodipino es más efectivo en el control de la PA en comparación
con nitrendipino, y presenta menos efectos adversos.
No obstante, el
efecto inmediato tras la primera dosis del nitrendipino comparado con un efecto
antihipertensivo gradual del amlodipino, esta baja inmediata de PA se compensa
con el aumento de la frecuencia cardíaca y aumento de efectos adversos como
cefalea y enrojecimiento.
ANALGESICOS
Los analgésicos son
uno de los grupos farmacológicos de mayor consumo en España: en 2007 los
analgésicos publicitarios (EFP) representaron el 14,5% de la cuota del mercado
EFP (se dispensaron un total de 19.528.007 unidades). La autora estudia los
distintos tipos de dolor, los analgésicos más relevantes y los consejos sobre
paliación del dolor que pueden ofrecerse desde la oficina de farmacia.

La palabra analgésico procede etimológicamente del
prefijo griego a-/an-(carencia, negación) y de algos (dolor). Según la
Asociación Internacional para el Estudio del Dolor (IASP), éste se define como
la experiencia sensorial y emocional desagradable asociada a una lesión hística
real o potencial, o descrita en términos de dicho daño. Por tanto, el dolor no
es solamente una sensación, es una experiencia. Además, no se trata únicamente
de algo sensorial, sino que también es algo emocional. En ocasiones, el dolor
no precisa de una lesión para manifestarse, puede existir en ausencia de ella.
El dolor es una experiencia subjetiva de gran
complejidad. Presenta un componente nociceptivo, responsable de la transmisión
de estímulos al sistema nervioso central que permiten defenderse ante
situaciones nocivas o peligrosas para el organismo, y un componente emotivo y
afectivo, que se caracteriza por irritabilidad, ansiedad y rabia en el caso de
un dolor agudo y que puede derivar incluso en depresión cuando se transforma en
un dolor crónico.
Los dos componentes del dolor son: sensorial y
emocional. El primero, que equivale a nocicepción (estimulación de las vías
nerviosas que conducen los estímulos dolorosos), se debe al estímulo de las
terminaciones sensoriales. Normalmente hay una relación entre el estímulo
sensorial y la intensidad del dolor (excepto en el dolor por inflamación o por
lesión nerviosa). El componente emocional equivale a la vivencia individual que
hace el paciente del estímulo nociceptivo. A menudo es el componente más
importante, especialmente en casos de dolor crónico.
Clasificación
En función de su duración, se diferencia entre el
dolor agudo y el crónico (tabla 1):
Tabla 1. Características de los dolores agudos y
crónicos
Dolor agudo
- Derivado
de una lesión tisular.
- Forma
parte de los mecanismos que protegen al organismo de su destrucción.
- Suele
desaparecer con la curación del proceso que lo causó.
- Ejemplos:
posoperatorio, dolor traumático, cefaleas tensionales, odontalgias y
lumbalgia aguda.
Dolor crónico
- Persiste
más allá de la curación de la lesión.
- A
menudo es difícil establecer una alteración hística que lo justifique.
- Se
deben considerar dolores crónicos aquellos que cursan con brotes durante
un largo período de tiempo.
- Ejemplos:
migrañas, dismenorreas, dolores osteoarticulares (artritis reumatoide,
artrosis, lumbalgia crónica), dolor neuropático (neuralgia del trigémino,
neuropatía diabética, neuralgia postherpética, distrofia simpática
refleja) y fibromialgia.
- Desde
el punto de vista fisiopatológico se distinguen varias fases:
- Dolor
de fase I o dolor fisiológico o nociceptivo. Se caracteriza por estímulos
nocivos breves y una buena correlación entre el estímulo y la intensidad.
Ejemplos: dolor de pinchadas leves y estímulos térmicos sin lesión.
- Dolor
de fase II o dolor inflamatorio. Se caracteriza por estímulos nocivos
persistentes y poca correlación entre el estímulo y la intensidad.
Ejemplos: dolor posoperatorio, dolor traumático y dolor reumático.
- Dolor
de fase III o dolor neuropático. Se caracteriza por una lesión nerviosa o
central y por la ausencia de correlación entre el estímulo y la intensidad
del dolor. Ejemplos: periférico (neuropatía diabética, síndrome del túnel
carpiano) y central (dolor talámico y distrofia simpática refleja).
FISIOPATOLOGIA DE
DOLOR
- Se
genera en las estructuras periféricas denominadas nociceptores.
- Viaja
al sistema nervioso central a través de las fibras aferentes primarias Aδ
y C.
- Se
procesa en el asta posterior de la médula espinal, donde se encuentra
sometido a influencias excitatorias e inhibidoras.
- Se
procesa en el encéfalo por vías específicas, principalmente la espinotalámica.
- Los
estímulos llegan al tálamo y posteriormente hasta la corteza
somatosensitiva.
- Existe
una modulación encefálica por diferentes áreas cerebrales.
- A
partir del mesencéfalo y de los núcleos del rafe existen vías inhibitorias
que se proyectan hacia la médula.
- El
estímulo persistente causa cambios plásticos en las neuronas nociceptivas
que podrían estar implicadas en el mantenimiento del dolor crónico.
TRATAMIENTO FARMACOLOGICO
- La
vía oral es siempre la de elección.
- Las
formas de liberación sostenida son útiles en el dolor crónico.
- Las
formas efervescentes y las líquidas suelen dar lugar a concentraciones
plasmáticas adecuadas con más rapidez que las formas sólidas.
- Se
debe recordar que la tolerancia oral de los AINE mejora con la ingesta de
alimentos y reduce los trastornos gastrointestinales.
- La
vía parenteral es útil en caso de intolerancia oral o cuando sea preciso
un efecto rápido.
- La
vía rectal presenta una absorción más inexacta en comparación con otras
vías.
- La
vía tópica basada en el uso de parches es conveniente en el dolor crónico.
- En
el caso de los AINE tópicos, se puede considerar que su eficacia es
limitada y depende de la correcta realización del masaje.
LOS ANALGESICOS SE CLASIFICAN EN PRIMMARIOS, SECUNDARIOS Y COADYUVANTES.
ANALGESICOS PRIMARIOS
Los analgésicos primarios tienen como efecto
farmacológico principal aliviar el dolor. Son de amplio espectro, es decir,
útiles en numerosos tipos de dolor. Se distinguen tres tipos:
- Analgésicos-antitérmicos
puros: paracetamol.
- Analgésicos-antiinflamatorios
(AINE): ácido acetilsalicílico (AAS), ibuprofeno, iCOX selectivos
(celecoxib y rofecoxib). Los AINE actúan bloqueando el enzima
ciclooxigenasa (COX) e impidiendo la síntesis de prostanoides
(prostaglandinas y tromboxanos). La COX tiene tres isoenzimas: COX-1,
COX-2 y COX-3. LOS AINE bloquean de forma diferencial los tres isoenzimas
lo que condiciona sus propiedades farmacológicas.
- Opioides:
agonistas puros de los receptores opioides (morfina, codeína, metadona,
fentanilo), parciales (buprenorfina), agonistas-antagonistas (pentazocina)
y mixtos (tramadol). Efectos indeseables:
- Frecuentes:
náuseas, vómitos, estreñimiento, sedación, miosis y picor.
- Se
presenta tolerancia por: analgesia, náuseas, vómitos, sedación y depresión
respiratoria.
- No
se presenta tolerancia por: miosis y estreñimiento.
Debido a estos efectos indeseables, se recomienda
la administración de antieméticos al inicio del tratamiento y de laxantes en
tratamientos de media y larga duración.
Se debe recordar
que la tolerancia oral de los AINE mejora con la ingesta de alimentos y así se
consigue reducir los trastornos gastrointestinales
ANALGESICOS SECUNDARIOS
Los analgésicos secundarios tienen otras
indicaciones, pero pueden disminuir algún tipo de dolor específico. Son de
espectro reducido y los hay de varios tipos:
- Antidepresivos:
amitriptilina y clorimipramina.
- Antiepilépticos:
carbamazepina, lamotrigina,
- Relajantes
musculares: diazepam, tetrazepam y gabapentina y topiramato. Están
indicados en el ciclobenzaprina (antidepresivo tricíclico empleado
tratamiento del dolor que tiene un origen como relajante muscular).
neurógeno o neuropático. El mecanismo
- Anestésicos
locales: lidocaína y prilocaína. fundamental de acción de los
antiepilépticos, en los síndromes neurógenos, es su acción sobre el sistema
Los coadyuvantes o auxiliares se administran de propagación de la descarga
neuronal, ya que al conjuntamente con los analgésicos primarios y/o
bloquear los canales de Na dependientes del voltaje secundarios para
potenciar sus efectos analgésicos o estabilizan la membrana neuronal y
suprimen la para contrarrestar sus efectos indeseables. Se emplean:
hiperexcitabilidad neuronal anómala.
- Corticoides.
- Psicofármacos:
neurolépticos, ansiolíticos y anfetaminas.
- Vasodilatadores:
antagonistas del calcio.
- Corticoides.
- Vasoconstrictores:
derivados ergóticos.
- Antieméticos
y laxantes.
LA ESCALERA ANALGESICA DE LA OMS

Se diseñó inicialmente para combatir el dolor
oncológico. Sin embargo, en la actualidad se utiliza en la mayoría de los
pacientes que presentan dolor. Consiste en una graduación progresiva de la
terapéutica analgésica de acuerdo con la respuesta obtenida por el paciente.
Implica el empleo inicial de paracetamol y AINE, para pasar después a las
asociaciones con opioides menores (codeína, etc.) y posteriormente a los
opioides mayores (morfina, etc.).
CONSEJOS DESDE LA FARMACIA
La elección de un analgésico para el tratamiento de
la sintomatología dolorosa debe realizarse siempre de modo individualizado. Es
necesario preguntar al paciente una serie de cuestiones que permitirán
seleccionar el medicamento publicitario más adecuado y proporcionar consejo
sanitario. Por ejemplo: ¿Qué síntomas presenta?
- ¿Cuándo
comenzaron?
- ¿Dónde
se localizan?
- ¿Cuál
es su intensidad?
- ¿Está
tomando algún medicamento?
- ¿Tiene
alguna enfermedad?
En relación con la posología conviene:
- Establecer
con claridad la posología eficaz y la máxima posible.
- Considerar
la administración a demanda o pautada.
- Determinar
el intervalo de administración.
- Aconsejar
cuándo y cómo tomar los analgésicos.
- Supervisar
la duración del tratamiento. Respecto a las interacciones farmacológicas,
el consejo farmacéutico debe ir encaminado a:
- Averiguar
los fármacos de prescripción y los de autoprescripción (incluidos los
productos fitoterapéuticos) que toma el paciente.
- Desaconsejar
la administración de medicamentos sin control médico previo.
- Desaconsejar
totalmente el consumo de alcohol conjuntamente con la toma de opioides.
Sobre las contraindicaciones, desde la oficina de farmacia habrá que:
- Asegurarse
de que las condiciones fisiológicas o patológicas del paciente no son una
limitación al tratamiento.
- Aconsejar
sobre las situaciones de precaución (conducción de vehículos o
manipulación de máquinas peligrosas).
- Determinar
si existen antecedentes que puedan restringir su uso (alergias). El
farmacéutico también puede orientar sobre las precauciones de uso:
- Aconsejar
cómo, cuándo y durante cuánto tiempo deben tomarse los medicamentos.
- Supervisar
el establecimiento de medidas o medicación coadyuvante (protectores
gástricos, antieméticos, laxantes, etc.)
- Detectar
la presencia de reacciones adversas o de falta de respuesta terapéutica.
Finalmente, es conveniente que desde la farmacia se detecten las
situaciones que aconsejan la visita médica, se deriven los pacientes al
médico cuando se sospecha la existencia de enfermedad grave, evitar la
automedicación y que los padres mediquen por su cuenta a los niños
pequeños.
ANALGESICOS RELEVANTES
Se describen a continuación los detalles sobre
dosificación, posología y precauciones de los analgésicos de uso más habitual.
- Ácido
acetilsalicílico (AAS) y derivados (salicilatos). La dosis oral para
adultos es de 500 mg cada 4 h. No se debe administrar en caso de úlcera
duodenal, asma, problemas de coagulación sanguínea, últimas semanas de
embarazo, niños menores de 12 años con infección vírica o lactantes (se
excreta en la leche materna).
- Paracetamol.
No es un AINE y es el analgésico más utilizado en el mundo. En los
territorios de influencia anglosajona se conoce como acetaminofeno. La
dosis por vía oral en adultos es de 325-650 mg cada 4 h. La dosis oral
infantil es de 20-40 mg/kg/día. Es de elección en pacientes que siguen un
tratamiento anticoagulante, ya que no altera la síntesis de protrombina ni
la agregación plaquetaria, así como en ulcerosos y en pacientes renales.
Es el analgésico y antipirético infantil ya que no causa síndrome de Reye.
No se debe administrar en caso de insuficiencia hepática y/o renal y no se
deben superar los 4 g diarios.
- Ibuprofeno.
En adultos, por vía oral, la dosis más frecuente es de 600-1.200 mg
repartidos en varias tomas, y no se recomienda superar los 1.200 mg
diarios. La dosis oral infantil es de 10 mg/kg/6 h con las comidas. No se
debe administrar a pacientes que presenten úlcera duodenal, asma,
problemas de coagulación o en el último trimestre de embarazo.
- Metamizol.
Es un derivado pirazolónico que actúa impidiendo la formación de
prostaglandinas, ya que inhibe la enzima ciclooxigenasa. Es analgésico y
antipirético, con un débil efecto antiinflamatorio. Está indicado en el
tratamiento de dolor agudo. La dosis oral en adultos es de 575 mg cada
6-8-12 h. La dosis parenteral suele ser de 2 g cada 8 h. No se debe
administrar en los siguientes casos: porfiria (trastorno del metabolismo
de los pigmentos sanguíneos que forman parte de la hemoglobina),
deficiencia de la enzima glucosa 6-fosfatodeshidrogenasa y antecedentes de
anemia aplástica o agranulocitosis (disminución del número de glóbulos
blancos en sangre).
- Tramadol.
Es un fármaco agonista de los receptores opioides que también actúa sobre
las vías serotonínicas. De acción central, actúa sobre las células
nerviosas específicas de la médula espinal y del cerebro. Está indicado en
el tratamiento del dolor de intensidad moderada a severa. La dosis oral en
adultos suele ser de 50 a 100 mg cada 6-8 h. La forma retard se inicia con
tratamientos de 50-100 mg cada 12 h y se puede aumentar hasta 150-200 mg
cada 12 h, dependiendo de la intensidad del dolor. La dosis inyectable en
adultos es inicialmente de 100 mg, pudiéndose administrar durante la hora
posterior 50 mg cada 10-20 min, sin sobrepasar una dosis total de 250 mg.
Posteriormente de 50 a 100 mg cada 6-8 h. No se debe administrar en casos
de insuficiencia hepática grave y de epilepsia no controlada con
tratamiento.
PROTECTORES DE LA MUCOSA GASTRICA
La
utilización de fármacos gastro protectores tanto en atención primaria como en
especializada ha aumentado considerablemente en los últimos años. Este
incremento, lleva aparejado en muchas ocasiones una utilización incorrecta de
estos fármacos según las directrices establecidas por las Sociedades
Científicas y los Comités de Farmacia y Terapéutica de los hospitales.
Las secreciones producidas por el estómago se pueden clasificar en dos
grupos: por un lado, los factores defensivos de la mucosa (moco y bicarbonato,
los cuales constituyen una capa gelatinosa firme protectora de la mucosa). Por
otro lado, los factores agresivos para la mucosa (ácido clorhídrico y
pepsinógeno).
En condiciones normales existe un equilibrio entre ambos tipos de
secrecciones de modo que se protege la integridad de la mucosa gástrica frente
a agentes externos como el alcohol y la bilis, o internos como alteraciones en
la función secretora de ácido que puede originar diversas enfermedades del
aparato digestivo.
LA SECRECCIÓN GÁSTRICA
Las células parietales del estómago secretan aproximadamente unos 2 l de
una solución isotónica de ácido clorhídrico 0,16 M. El mecanismo de producción
de la secrección ácida consiste en la síntesis dentro de la célula parietal de
ácido carbónico por acción de la anhidrasa carbónica. Este ácido carbónico se
disocia en ión bicarbonato (HCO3-) e ión hidrogenión (H+), el HCO3- se
intercambia a través de la membrana basal por Cl-, que sale, junto con el K+
hacia la luz del estómago. El K+ se intercambia por H+ del interior de la
célula mediante una ATPasa K+/H+, originándose un acúmulo de H+ en la luz del
estómago.
La secrección ácida es regulada por varias sustancias que interaccionan
con receptores específicos de la célula y actúan como primeros mensajeros.
Estas sustancias son: histamina, acetilcolina y gastrina.
La histamina interactúa con los receptores H2 de la membrana de la
célula parietal activando la enzima adenilciclasa en el interior celular, la
cual transforma el ATP citosólico en AMPcíclico (AMPc). El AMPc a su vez
estimula proteincinasas que fosforilan proteínas que actúan como mediadores en
la síntesis de hidrogeniones y su posterior secrección hacia la luz del
estómago.
La acetilcolina estimula los receptores muscarínicos de la superficie de
las células parietales y de los mastocitos.
La gastrina actúa sobre receptores específicos de la célula parietal
provocando la secrección directa de ácido.
LA BARRERA MUCOSA
La barrera mucosa engloba todos los factores que participan en el
mantenimiento de la integridad de la mucosa en el medio ácido estomacal. Los
factores que contribuyen a su estabilidad son los siguientes:
La secrección de moco y bicarbonato. El moco es un gel viscoso
constituido por glucoproteinas que forman una capa de 2 cm que recubre la
superficie de la mucosa. Su misión es proteger la integridad de las células de
la superficie, lubricar la mucosa e hidratarla reteniendo agua. La secrección
de bicarbonato se origina mediante el intercambio de Cl-/HCO3- en la membrana
luminal de las células epiteliales. Por cada ión H+ secretado por la célula
parietal, una molécula de CO2 procedente de la circulación sanguínea se
convierte en bicarbonato de modo que el bicarbonato queda retenido en el
interior de la capa de moco y neutraliza la retrodifusión de H+. La capa de
moco-bicarbonato crea un gradiente de pH desde el medio ácido luminal hasta la
neutralidad de la superficie de la mucosa.
Flujo sanguíneo de la mucosa gástrica. Este flujo contribuye a
satisfacer las necesidades metabólicas requeridas por los diferentes procesos
secretores o de reparación de la mucosa y arrastra el ácido que ha difundido a
través del epitelio. El aumento de flujo sanguíneo es un mecanismo defensivo
frente a agentes lesivos a nivel tópico. La disminución del flujo origina el
mecanismo primario de la lesión de la mucosa gástrica por algunos agentes
ulcerogénicos.
Restitución celular. Es el
mecanismo inicial de reparación de la mucosa ante una lesión aguda causada por
agentes tópicos. Mediante este proceso es posible reparar el epitelio
superficial dañado en un período de 30 min a 4 h.
Consiste en la migración de las células próximas a las zonas donde las
células lesionadas se han exfoliado. Para que sea efectivo se requiere un flujo
sanguíneo adecuado. La restitución se inhibe con la presencia masiva de ácido.
Los factores agresivos más frecuentes que pueden comprometer la
resistencia de la barrera mucosa son la infección por Helicobacter pylori y los antiinflamatorios no
esteroideos (AINE).
EL GRUPO DE LOS ANTIÁCIDOS ESTÁ INTEGRADO POR COMPUESTOS INORGÁNICOS QUE
REACCIONAN CON EL HCL GÁSTRICO, CON LO QUE REDUCEN LA ACIDEZ GÁSTRICA Y
ADSORBEN LA PEPSINA Y OTROS ENZIMAS PROTEOLITICOS, DISMINUYENDO LA AGRESIVIDAD
QUÍMICA Y ENZIMÁTICA SOBRE LA MUCOSA
CONSEJOS
DESDE LA OFICINA DE FARMACIA
En el caso de la enfermedad gástrica, la atención farmacéutica debe
prestarse en función de los síntomas y del medicamento prescrito. Así, entre
los consejos generales podemos destacar los siguientes:
El tratamiento debe seguirse
durante el período establecido por el médico, aunque los síntomas remitan.
Se debe consultar al médico si los síntomas continúan o empeoran al cabo
de varios días de tratamiento, notificando especialmente síntomas como
sensación de saciedad, debilidad o pérdida de peso injustificada.
Si el paciente experimenta
síntomas como dispepsia, disfagia, sensación de saciedad, síntomas de
hemorragia gástrica, como melena (deposición negra, brillante y viscosa, de
olor fétido, semejante al alquitrán, constituida por sangre coagulada y
digerida, sola o mezclada con heces), hematemesis (vómito de sangre procedente
de las partes altas del tubo digestivo), anemia o pérdida de peso injustificada,
se deberá descartar la existencia de carcinoma esofágico o gástrico antes de
administrar estos medicamentos.
El misoprostol no debe ser utilizado por mujeres embarazadas (riesgo de
aborto y/o hemorragia uterina). En mujeres en edad fértil, se aconseja adoptar
métodos anticonceptivos eficaces, y si la paciente quedase embarazada,
suspender el tratamiento. Se aconseja realizar una prueba de embarazo antes de
iniciar la terapia.
Se aconseja tomar los inhibidores de la bomba de protones por las
mañanas. Si se requiere una segunda dosis, se tomará una hora antes de la cena.
Se deben ingerir las cápsulas y los comprimidos enteros sin masticarlos,
partirlos o pulverizarlos. Para pacientes con dificultades para tragar, los
comprimidos pueden dispersarse en medio vaso de agua.
ENFERMEDADES
ASOCIADAS AL EXCESO DE SECRECCIÓN ÁCIDA
GASTRITIS
Se denomina gastritis a la inflamación aguda o crónica de la mucosa
gástrica. Se pueden dividir los tipos de gastritis en tres grupos:
En el primer grupo se encuentran
las gastritis, con clara expresión macroscópica con erosiones y/o hemorragia,
que corresponden a inflamaciones agudas de la mucosa estomacal.
El segundo grupo corresponde a las inflamaciones de la mucosa
histológicamente demostradas con alteraciones mínimas inespecíficas que suelen
corresponder a procesos inflamatorios crónicos e inespecíficos del estómago o a
fenómenos inflamatorios acompañantes de afecciones concretas, como la úlcera
gástrica.
El tercer grupo engloba todas las
inflamaciones específicas que corresponden a enfermedades bien caracterizadas,
ya sea limitadas al estómago o formando parte de un proceso inflamatorio
específico y difuso del tubo digestivo.
EN LA PRÁCTICA CLÍNICA SE RECOMIENDA VALORAR EL BENEFICIO-RIESGO DE LOS
IBP EN GRUPOS COMO LOS PACIENTES ANCIANOS CON EPOC QUE TOMAN INMUNOSUPRESORES O
CORTICOIDES DE FORMA CRÓNICA y A MENUDO ANTIBIÓTICOS
ÚLCERA PÉPTICA
La úlcera péptica se origina como consecuencia del desequilibrio entre
los factores agresivos y defensivos de la mucosa gástrica y/o duodenal. La
importancia de la secrección ácida y de la actividad péptica en la patología de
la úlcera péptica es fundamental ya que, en ausencia de ácido, no hay úlcera. Además
se aprecia una relación directa entre la eficacia del tratamiento antisecretor
en la cicatrización de la úlcera y la supresión de la acidez gástrica.
ESOFAGITIS POR REFLUJO GASTROESOFÁGICO
El tránsito del contenido gástrico hacia el esófago no siempre se
considera patológico. En circustancias normales la frecuencia de episodios de
reflujo es mínima, el tiempo de contacto del ácido gástrico con la mucosa
esofágica es corto y la resistencia del esófago frente a agentes agresivos es
adecuada. Algunos alimentos como las comidas grasas, el chocolate, el alcohol,
el tabaco y la menta disminuyen la presión del esfínter esofágico inferior y
facilitan el paso del contenido gástrico al esófago.
La aparición de episodios de regurgitación ácida, que aumentan con la
ingesta y con el decúbito, y se alivian con agentes alcalinos, es indicativa de
reflujo gastroesofágico. No obstante, los síntomas del reflujo pueden ser muy
variados: eructos, dolor epigástrico, pesadez posprandial, náuseas, hipo,
disfagia, odinofagia (sensación dolorosa al tragar alimento), molestias
faríngeas y/o laríngeas, afonía, alteraciones respiratorias o dolor torácico.
Si bien la fisiopatología de la esofagitis está directamente relacionada
con la motilidad gastrointestinal, la mayoría de los síntomas que presenta son
debidos al daño que el reflujo ácido-péptico origina sobre el epitelio del
esófago. Por ello, el tratamiento se basa en la eliminación del ácido. Dentro
de los fármacos antisecretores, los inhibidores de la bomba de protones son los
más eficaces para la curación de la enfermedad. En aquellos casos en los que la
sintomatología clínica así lo requiera, como, por ejemplo, en episodios
puntuales de acidez, se puede complementar el tratamiento con un antiácido
pautado a demanda.
SÍNDROME DE ZOLLINGER-ELLISON
El síndrome de Zollinger-Ellison se caracteriza por la presencia de
hipersecrección ácida, enfermedad ulcerosa grave del tracto digestivo superior
y tumor generalmente pancreático, de células no beta, secretor de gastrina. Los
pacientes afectados presentan cantidades elevadas de gastrina sérica circulante
proveniente del tumor. La enfermedad cursa con síntomas relacionados con las
úlceras pépticas. Es frecuente que estas úlceras se compliquen con hemorragia o
perforación y que coexistan con enfermedad esofágica péptica, con esofagitis,
úlceras esofágicas y/o estenosis. Los pacientes también pueden presentar
síntomas como diarrea, esteatorrea y pérdida de peso. El tratamiento se basa en
controlar la úlcera y resecar el tumor productor de gastrina o paliar los
efectos de su diseminación.
FÁRMACOS
ANTISECRETORES
ANTAGONISTAS
DE LOS RECEPTORES H2
Dentro de este grupo se engloban los siguientes medicamentos:
cimetidina, ranitidina, famotidina, nizatidina y roroxatidina, todos ellos se
caracterizan por presentar acciones antiúlcera péptica, ser antisecretores
gástricos y antagonistas específicos, competitivos y reversibles de los
receptores de histamina H2 situados en las células parietales gástricas. Los
receptores H2 al ser estimulados activan una adenilatociclasa, causando un
aumento de los niveles de AMPc, que por un mecanismo en cascada, en la que
interviene la fosforilación de distintas proteínas, activa la bomba de protones
y, por tanto, la secrección ácida gástrica. Son antagonistas muy específicos y
no afectan a los receptores H1 ni presentan efectos anticolinérgicos. Asimismo,
inhiben la secrección gástrica basal, la estimulada por cafeína, histamina,
gastrina, agonistas colinérgicos, alimentos, insulina y la secrección nocturna.
No afectan al vaciado gástrico ni a la presión del esfínter esofágico inferior.
En cuanto a las interacciones medicamentosas, los antagonistas H2,
especialmente la cimetidina, interfiere con los medicamentos que se metabolizan
a través de la vía hepática del citocromo P450, concretamente la cimetidina
interfiere con las enzimas CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 y CYP3A4. Al
reducir la capacidad de metabolizar ciertos fármacos que dependen de estas
enzimas, se produce un aumento de las concentraciones de éstos en el plasma
sanguíneo que en ocasiones pueden llegar a ser tóxicas. Algunos de los fármacos
afectados son: warfarina, teofilina, fenitoína, lidocaína, quinidina,
propranolol, labetalol, metoprolol, antidepresivos tricíclicos, algunas
benzodiazepinas, sulfonilureas, metronidazol, etanol y MDMA.
INHIBIDORES DE LA BOMBA DE PROTONES
En la categoría de los inhibidores de la bomba de protones (IBP) se engloban
los siguientes fármacos: esomeprazol, lansoprazol, omeprazol, pantoprazol y
rabeprazol, todos ellos caracterizados por su acción antiúlcera péptica, de
antisecretores gástricos e inhibidores específicos de la bomba de protones
ATPasa H+/K+ de la célula parietal gástrica, por lo que impiden la producción
de ácido gástrico inducida tanto por acetilcolina como por gastrina o histamina
(tabla 2).
Son profármacos que deben activarse en la luz de los canalículos
secretores de las células parietales para ser activos. Se comportan como bases
débiles que, tras su absorción, se distribuyen por el organismo. Al llegar a
las células parietales, en un medio muy ácido, sufren una transposición
molecular, dando lugar a la forma activa, un derivado sulfonamido hidrófilo,
por lo que no puede acceder de nuevo al torrente circulatorio y se acumula en
ellas. La inhibición de la ATPasa se debe a la formación de puentes disulfuro
con residuos de cisteína de la cadena alfa luminal de la bomba de protones.
Esta inhibición es prácticamente irreversible, no competitiva y dependiente de
la dosis.
La célula parietal sólo recupera la actividad secretora mediante la
síntesis de nuevas moléculas de la bomba, por este motivo la duración de los
efectos de estos fármacos puede ser de hasta 4 días tras la administración de
una dosis única.
FÁRMACOS
ANTIÁCIDOS
El grupo de los antiácidos está integrado por compuestos inorgánicos que
reaccionan con el HCl gástrico, con lo que reducen la acidez gástrica, adsorben
la pepsina y otros enzimas proteolíticos, disminuyendo la agresividad química y
enzimática sobre la mucosa digestiva, especialmente en las zonas donde ésta se
encuentra deteriorada. Los antiácidos disminuyen el dolor ulceroso, pero no
favorecen la cicatrización ni impiden las recaídas. En la tabla 3 se recogen
las indicaciones y los medicamentos correspondientes a este grupo.
FÁRMACOS PROTECTORES DE LA MUCOSA
Los fármacos protectores de la mucosa son los que como su nombre indica
protegen la mucosa del tracto gastrointestinal de la secrección ácida y de las
enzimas digestivas, además presentan acción antiúlcera péptica y antisecretora
gástrica. En la tabla 4 se recoge el mecanismo de acción principal, las
indicaciones y los medicamentos comercializados.
ABUSO DE PROTECTORES GÁSTRICOS
Los protectores gástricos son fármacos que se prescriben frecuentemente
tanto en atención primaria como en especializada. En la mayoría de estudios
realizados sobre utilización y evaluación de la prescripción de estos
medicamentos, se ha detectado que esta prescripción no siempre se ajusta a las
indicaciones de la ficha técnica, por lo que según el RD 1015/2009, de 19 de
junio, que regula la disponibilidad de medicamentos en situaciones especiales,
se consideraría como utilización en condiciones diferentes a las
autorizadas (off-label). El médico prescriptor debería justificar en la
historia clínica la necesidad del uso del medicamento, informar al paciente de
posibles beneficios y riesgos potenciales y obtener su consentimiento
informado.
Asimismo, en estudios realizados en el medio hospitalario, se ha
detectado que la prescripción de estos medicamentos tampoco se ajusta a las
recomendaciones establecidas por los Comités de farmacia y terapéutica. Se ha
relacionado el empleo de estos fármacos con la profilaxis de la úlcera de
estrés y la profilaxis de sangrado digestivo sin tener asociados estos
pacientes otros factores de riesgo. Un estudio realizado por Hermida Ameijeiras
y colaboradores sobre la prevalencia de prescripción-indicación de protectores
gástricos en medio hospitalario detectó una prevalencia de prescripción
injustificada de gastroprotectores del 64,8%, de la cual el 96,3% se debió a
prescripciones de IBP. Este estudio destaca la amplia utilización de los IBP en
terapia preventiva para pacientes con tratamiento corticoideo no asociado a
AINE. En otros estudios, además de obtenerse resultados semejantes, también se
ha concluido que el 50% de los pacientes con tratamiento injustificado siguen
recibiéndolo de forma ambulatoria hasta 3 meses más tarde, con lo que ello
implica respecto a las interacciones medicamentosas, los efectos adversos y el
incremento del gasto sanitario.
A continuación se exponen algunas de las consecuencias relacionadas con
la sobreutilización de los IBP por ser este grupo de medicamentos el más
ampliamente utilizado dentro de los protectores gástricos.
Los IBP son fármacos muy seguros, pero en los últimos años se ha
relacionado su utilización crónica con un aumento en el número de infecciones
entéricas e infecciones por Clostridium difficile,
déficit de absorción de iones calcio, iones hierro y vitamina B12 y un aumento
en el riesgo de fracturas de cadera y de neumonía adquirida en la comunidad.
Está demostrado que para la absorción correcta del calcio es necesario
el medio ácido del estómago y del duodeno proximal. Una alteración en la
absorción de calcio podría originar hiperparatiroidismo secundario, activándose
mecanismos compensatorios como la reabsorción osteoclásica que puede conducir a
la disminución de la masa ósea y, por tanto, al aumento del riesgo de
fracturas. Si la producción del ácido gástrico es necesaria para la ionización
del calcio y su posterior absorción, puede esperarse en teoría que un uso
crónico de IBP dé lugar a un aumento del riesgo de fracturas. Sin embargo, no
hay estudios bien definidos que relacionen ambos factores.
El consumo crónico de estos fármacos también se ha asociado a un aumento
de la incidencia de neumonía adquirida en la comunidad. En la práctica clínica
se recomienda valorar el beneficio-riesgo en pacientes de riesgo como pueden
ser los ancianos con EPOC que toman inmunosupresores o corticoides de forma
crónica y a menudo antibióticos.
La sobreutilización de estos medicamentos es un aspecto que debe
controlarse para evitar los efectos adversos que se están detectando, por ello
las dosis, la duración del tratamiento y las indicaciones deberían ajustarse
estrictamente a lo establecido en su ficha técnica hasta que se disponga de
datos concluyentes obtenidos mediante la realización de estudios prospectivos
bien diseñados que puedan medir el efecto de estos inhibidores en los efectos
descritos.
ANTICUAGULANTES
Los anticoagulantes
son un grupo de sustancias de distinta naturaleza química relacionados por su
efecto biológico. Se pueden dividir en:
Anticoagulantes de
acción directa: aquellos que por sí solos son capaces de inhibir la cascada de
la coagulación. Ejemplos: inhibidores directos de trombina (hirudina,
argatroban).
Anticoagulantes de
acción indirecta: aquellos que mediante su interacción con otras proteínas o
actuando en otras vías metabólicas, alteran el funcionamiento de la cascada de
la coagulación. Ejemplos: inhibidores mediados por antitrombina III (heparina
no fraccionada, heparinas de bajo peso molecular, danaparoide sódico);
inhibidores de la síntesis de factores de coagulación (derivados del
dicumarol).
Pueden administrarse
por vía parenteral (subcutánea o endovenosa) para inducir un estado
hipocoagulante en forma rápida. En clínica esta ruta se usa, habitualmente, por
cortos períodos de tiempo. Cuando se administran por vía oral el efecto
anticoagulante, es de lenta instalación. En general, esta vía es utilizada en
los tratamientos de mantención.
HEPARINA NO FRACCIONADA (HNF)
Es una mezcla de
glicosaminoglicanos extraída del cerdo o bovino, con una variable número de
residuos que les dan cargas negativas. Existen formas comerciales con pesos
moleculares entre 5 y 30 kd (media 15 kd).
MECANISMO DE ACCION
Se une a antitrombina
III (ATIII), produciendo un cambio conformacional que aumenta la capacidad
inhibitoria de esta enzima sobre los factores de coagulación: trombina, Xa y
IXa. Para que la inactivación de trombina sea acelerada debe formarse un
complejo terciario de ATIII + heparina + trombina. El factor Xa sólo requiere
del cambio conformacional.
La limitación
biológica de la reacción está determinada por la incapacidad del complejo
ATIII+heparina de inhibir al factor Xa y a la trombina que ya están unidas al
coágulo
FARMACOCINETICA
La vida media de la heparina depende del tamaño de las
moléculas y de la dosis administrada. Su depuración ocurre por depolimerización
intracelular, siendo las moléculas más grandes las que más rápido se depuran.
El sistema es saturable, de modo que una dosis de 100 UI/kg en bolo e.v. es
depurada en 1 hora, mientras una de 25 UI/ kg sólo en media hora. Sus cargas
negativas la unen de forma inespecífica a diversas proteínas plasmáticas
(vitronectina, fibronectina, lipoproteínas, fibrinógeno, factor plaquetario 4
[PAF4], factor von Willebrand), lo que reduce el número de moléculas de
heparina disponibles para combinarse con ATIII. La concentración de estas
proteínas ligadoras inespecíficas es variable de una persona a otra. Este hecho
explica la variabilidad del efecto anticoagulante obtenido a dosis iguales en
personas diferentes.
CONTROL DE TERAPIA
Las limitaciones farmacocinéticas expuestas obligan a
realizar un control estricto de la terapia, para evitar la sobre o sudorificación.
Esto es válido cuando la heparina es utilizada a dosis terapéuticas, buscando
anticoagulación inmediata. Cuando se utiliza a dosis bajas, como prevención de
tromboembolismo venoso, no se requiere de control.
Se pueden
monitorizar directamente los niveles plasmáticos de heparina por titulación con
protamina o midiendo la actividad anti-Xa del suero heparinizado. Si se
utilizan estas técnicas se recomienda mantener niveles plasmáticos de 0,2 a 0,4
UI/ml con la primera, y de 0,3 a 0,6 UI/ ml con la segunda. En clínica lo más
frecuente es utilizar una prueba de coagulación funcional, como el TTPa, que
mide los 3 factores inhibidos. Esto último debe acompañarse de la
estandarización del TTPa por el laboratorio local, ya que los diferentes tipos
de reactantes y equipos de medición disponibles, arrojan diferentes tiempos de
TTPa para una misma concentración plasmática de heparina. El concepto
“resistencia a la heparina” se emplea para designar a aquellos casos en que,
pese a dosis crecientes de heparina, no se observa la esperada prolongación del
TTPa o ésta es insignificante. En dichos casos lo más recomendable es solicitar
la medición de los niveles de heparina, antes de seguir subiendo las dosis.
REACCIONES ADVERSAS
1. Asociadas a sobredosis: Sangrado. Si el sangrado es
leve basta con suspender la infusión por 1 hora, y reiniciar con una dosis más
baja. Si esta complicación amenaza la vida, puede usarse el antagonista Sulfato
de Protamina (1 mg neutraliza 100 UI de heparina). El cálculo de la dosis de
protamina requerida para antagonizar toda la heparina administrada, puede
efectuarse sumando el total pasado en 1 hora, más la mitad de la dosis de la
hora previa, más un cuarto de la dosis pasada 3 horas antes. Ejemplo: Para una
infusión de 1500 U/hora el cálculo es: 1500UI+750UI+ 375UI =2625UI de heparina
a neutralizar =26,25 mg de protamina a pasar e.v. en 30 a 60 min. Los
diabéticos en tratamiento con insulina NPH[lenta] pueden presentar anafilaxia.
2. Asociadas a uso prolongado: Osteoporosis. La heparina
induce reabsorción ósea acelerada. En general se observa luego de 3 meses de
uso.
3. Asociadas a
formación de complejos inmunes: Síndrome de trombocitopenia / trombosis y
necrosis cutánea por heparina. La unión de heparina con el factor plaquetario 4
(PAF4) puede inducir la formación de autoanticuerpos. Los complejos inmunes
(PAF4-heparina-IgG), son capaces de activar a las plaquetas provocando un
estado de hipercoagulabilidad paradójico, con consumo plaquetario
(trombocitopenia), y coagulación intravascular (trombosis y necrosis cutánea por
isquemia). El cuadro se desarrolla luego de 5 dias de tratamiento con heparina
en hasta el 5% de los pacientes, y puede evitarse acortando el tiempo de su
utilización al imbricar precozmente la infusión de heparina con los anticoagulantes
orales.
4.Asociada a impurezas en la mezcla Urticaria.
VIAS DE ADMINISTRACION POSOLOGIA
Cuando la heparina no fraccionada se usa a dosis
terapéuticas, se recomienda la via endovenosa continua con bomba de infusión.
En este caso la posologia debe ser ajustada según el peso del paciente
iniciando con un bolo 0.v. de 80 Ul/kg de peso, y luego con la infusión
continua de heparina a 18 Ul/kg/hora
La dosis a infundit debe corregirse periódicamente, ya
sea directa (niveles plasmáticos) o indirectamente (TTPa), de modo que el
paciente mantenga un nivel de anticoagulación dentro del rango terapéutico.
Cada contro debo confeccionar nomogramas do corrección de dosis ajustados a sus
valores de TTPa estandarizados, que faciliten el uso del medicamento. Los
controles so realizan a intervalos de 6 horas, que corresponde al tiempo
requerido por la infusion para alcanzar un estadode cinética estable.
Cuando la heparina es usada a dosis profilácticas se
recomienda la via subcutánea. con intervalos de administración de 8 a 12 horas,
debido a que la absorción es mucho más lenta que cuando se usa por vía
endovenosa. En esta situación no se requiere de control de laboratorio
USOS CLINICOS
Pueden usarse en la prevención o el tratamiento de
cuadros trombóticos
Prevención de tromboembolismo venoso:
1. En patologias médicas con factores de nesgo de
complicación con trombosis venosa (TVP) como cáncer, insuficiencia cardiaca,
enfermedad pulmonar crónica severa, infarto agudo al miocardio. accidente
vascular encefálico con parosia o parálisis, y en los enfermos postrados. En
estos casos se recomienda usar dosis de 5000UI c/12 horas via subcutánea.
2- En los pacientes con cirugia general con o sin
factores de riesgo, y aquellos con cirugia ginecológica o urológica sin factores
de riesgo de TVP, también se recomiendan 5000UI c/12 horas via subcutánea.
3.En los pacientes con cirugía ginecológica o urológica
que tienen factores de riesgo de desarrollar TVP (cáncer), se recomiendan
5000UI c/12 horas vía subcutánea asociado a métodos de prevención mecánicos
(compresión neumática intermitente de extremidades o vendaje elástico). Los
pacientes con cirugía ortopédica mayor (prótesis de cadera, prótesis de rodilla
o fractura de cadera), tienen una probabilidad de complicación con TVP significativamente
mayor, aun en ausencia de los factores de riesgo ya citados. Estos pacientes
deben ser protegidos con heparina de bajo peso molecular, ya que el uso de la
heparina no fraccionada, aun con medidas físicas asociadas, no reduce en forma
importante la ocurrencia de TVP.
Tratamiento de enfermedades tromboembólicas:
1.En la TVP se recomienda la infusión continua de
heparina para alcanzar un TTPa equivalente a niveles de heparina de 0,2 a
0,4UI/ml o de 0,3 a 0,6UI/ml, dependiendo si se usa la titulación con protamina
o la actividad anti-Xa respectivamente.
2. En la embolia
pulmonar aguda también se recomienda la infusión continua con heparina, para
alcanzar un TTPa equivalente a los niveles plasmáticos señalados para la TVP.
3. En la insuficiencia arterial aguda de extremidades
(embolia o trombosis), se recomienda la infusión continua con heparina con
igual meta de TTPa que en la TVP.
4. En la
prevención de embolización sistémica en el período pericardioversión de la
arritmia completa por fibrilación auricular o del flutter auricular, se
recomienda también la infusión continua con igual meta de TTPa que para la TVP.
5.En la angina inestable, se recomienda la infusión
continua con heparina para alcanzar un TTPa equivalente a los mismos niveles
plasmáticos que en la TVP.
HEPARINAS DE BAJO PESO MOLECULAR (HBPM)
La depolimerización química o enzimática de la heparina
no fraccionada produce moléculas más pequeñas con pesos moleculares entre 1 y
10kd (media kd), denominadas heparinas de bajo peso molecular. Dependiendo de
la técnica de depolimerización (fraccionamiento) utilizada, se obtienen
distintos tipos de HBPM, cuyas propiedades farmacocinéticas son también
distintas.
MECANISMO DE ACCION
Tal como la heparina no fraccionada (HNF), aceleran la
inhibición del factor Xa y la trombina por ATIII, con la que forman un
complejo. Sin embargo, se diferencian en que las HBPM inhiben más al factor Xa
que a la trombina (relación de inactivación Xa: trombina de 4:1 a 2:1). El
complejo HBPM+ATIII, al igual que el complejo HNF+ATIII, tampoco puede inhibir
al factor Xa que ya está unido al coágulo.
FARMACOCINETICA
Al ser más pequeñas que la heparina no fraccionada, las
HBPM se unen menos a células, depurándose más lento, se absorben mejor por vía
subcutánea y su unión a proteínas plasmáticas diferentes a ATIII es menor.
Lo anterior permite:
- Una mejor
relación dosis:respuesta. Es decir un efecto anticoagulante equivalente a igual
dosis, en personas diferentes.
- La
administración 1 ó 2 veces al día sin necesidad de control de laboratorio.
- Un tratamiento
ambulatorio seguro de pacientes con TVP no complicada.
Otras ventajas son:
- Producen menos
síndrome de trombocitopenia /trombosis y necrosis cutánea por heparina.
- Probablemente producen menos osteoporosis.
- Probablemente
producen menos sangrado.
TROMBOTICOS
|
En
los últimos años la terapia fibrinolítica se ha desarrollado más ampliamente
que otras áreas de la medicina. Cada año, las publicaciones sobre terapia
trombolítica van en aumento, debido a que, las enfermedades tromboembólicas
son una de las principales causas de morbimortalidad en el mundo occidental. El tratamiento fibrinolítico tiene como fin
potenciar la trombólisis, restaurando el flujo de un vaso (arterial o venoso)
ocluido recientemente por un trombo. Está dirigido al tratamiento del trombo
más que a la causa de la trombosis. Se diferencia por ello del tratamiento
anticoagulante, el cual se emplea primariamente para prevenir la formación de
trombos y evitar la progresión y extensión de los ya formados. Los fármacos fibrinolíticos son proteasas que
actúan como activadores directos o indirectos del plasminógeno, dando lugar a
la conversión de esta proenzima en su forma activa (plasmina), que a su vez
cataliza la degradación de fibrina o fibrinógeno y la disolución del coágulo.
Estos fármacos pueden subdividirse teóricamente en activadores
“fibrinespecíficos” y “no fibrinespecíficos”. Los activadores “no fibrinespecíficos” como la
estreptoquinasa (SK), la uroquinasa (UK), y la anistreplasa (APSAC),
convierten tanto al plasminógeno circulante como al unido al coágulo en
plasmina, dando lugar no sólo a la lisis de la fibrina en el coágulo, sino
también a una importante fibrinogenolisis sistémica, fibrinogenemia y
elevación de los productos circulantes de la degradación de la fibrina (PDF). En virtud de su relativa selectividad por el
complejo binario plasminógeno-fibrina, los activadores “fibrinespecíficos”
(t-PA, scu-PA, reteplasa) dan lugar, fundamentalmente, a la lisis de fibrina
en la superficie del coágulo sin afectar teóricamente al fibrinógeno
circulante (31). Los fármacos trombolíticos han sido clasificados
también como de primera, segunda y tercera generación, según se han ido
incorporando a la terapéutica habitual de las enfermedades
tromboembólicas (tabla III). AGENTES
TROMBOLÍTICOS DE 1ª GENERACIÓN
|


No hay comentarios:
Publicar un comentario